
Эпигенетический подход: Результаты и перспективы генетики когнитивных нарушений при шизофрении: эпигенетические подходы
Результаты и перспективы генетики когнитивных нарушений при шизофрении: эпигенетические подходы
В клеточном ядре ДНК в комплексе с гистоновыми белками образует трехмерную структуру — хроматин, от которой зависит доступ транскрипционного аппарата клетки к ДНК. Эпигенетика изучает механизмы изменения состояния хроматина, не затрагивающие нуклеотидную последовательность ДНК, но влияющие на экспрессию генов — эпигенетические модификации [1]. В узком смысле ее предметом является метилирование ДНК и посттрансляционные модификации гистонов. Однако часто к сфере изучения эпигенетики относят любые локальные свойства хроматина, могущие влиять на активность генома.
Метилирование ДНК представляет собой присоединение метильной группы к основанию цитозина © и имеет место в подавляющем большинстве случаев в участках ДНК, где за цитозином следует гуанин (G), т. е. динуклеотидах CpG. Еще в 60-х годах прошлого века было установлено [2], что в геномах многих организмов CpG-динуклеотиды встречаются реже других видов. Более того, CpG-динуклеотиды распределены в геноме неслучайно. Часто они формируют обогащенные CpG-динуклеотидами компактные области (CpG-островки), расположенные в функционально значимых частях генома. Так, их находят в промоторах большинства генов в геноме человека. Эти особенности связаны с метилированием цитозинов [3]. В типичном случае локальное метилирование ДНК привлекает неспецифические метил-CpG-связывающие белки, что ведет к подавлению транскрипции соседних генов. Напротив, неметилированные сайты CpG в составе СpG-островков привлекают белки, препятствующие подавлению транскрипции. Посттрансляционные модификации определенных аминокислотных остатков гистонов на N-конце (ацетилирование, метилирование и др.) связаны как с активацией, так и с подавлением экспрессии генов [4, 5].
Более того, CpG-динуклеотиды распределены в геноме неслучайно. Часто они формируют обогащенные CpG-динуклеотидами компактные области (CpG-островки), расположенные в функционально значимых частях генома. Так, их находят в промоторах большинства генов в геноме человека. Эти особенности связаны с метилированием цитозинов [3]. В типичном случае локальное метилирование ДНК привлекает неспецифические метил-CpG-связывающие белки, что ведет к подавлению транскрипции соседних генов. Напротив, неметилированные сайты CpG в составе СpG-островков привлекают белки, препятствующие подавлению транскрипции. Посттрансляционные модификации определенных аминокислотных остатков гистонов на N-конце (ацетилирование, метилирование и др.) связаны как с активацией, так и с подавлением экспрессии генов [4, 5].
Метилирование ДНК, так же как и другие эпигенетические метки, отражает статус работы гена и может являться интегральным показателем различных средовых и генетически обусловленных сигналов на молекулярном уровне.
Результаты полногеномного изучения ассоциаций (GWAS), транскриптомные данные и эпигенетические исследования свидетельствуют, что эпигенетические модификации играют важную роль в развитии шизофрении [6, 7]. Большая часть ассоциаций с шизофренией, обнаруженных в GWAS, приходится не на кодирующие, а на регуляторные участки ДНК [8]. В частности, биоинформатический анализ результатов полногеномных и полноэкзомных исследований шизофрении [9, 10] указывает на устойчивую ассоциацию с шизофренией полиморфных локусов, связанных с метилированием гистонов. В близнецовых эпигенетических исследованиях [11—13] обнаружены различия в метилировании отдельных локусов ДНК в дискордантных по шизофрении монозиготных парах.
Особенности эпигенетических процессов при шизофрении позволяют предположить, что эпигенетические механизмы играют роль и в формировании сопутствующих заболеванию когнитивных нарушений. Кроме того, основанием для этого предположения являются данные о роли эпигенетических модификаций в собственно когнитивных процессах [17, 18] и вызванных стрессом изменениях строения и работы мозга [19—21].

В опытах на экспериментальных животных было выявлено действие эпигенетических механизмов, запускаемых средовыми факторами риска шизофрении, на когнитивные процессы и особенности работы мозга. Например, M. Labouesse и соавт. [29] обнаружили, что пренатальная инфекция (инфекция матери в период беременности) ведет к увеличению в префронтальной коре головного мозга уровня метилирования в промоторе гена
GAD1, связанного с синтезом γ-аминомасляной кислоты (ГАМК), что в свою очередь коррелирует с выраженностью вызванных пренатальной инфекцией нарушений рабочей памяти и социальных взаимодействий. J. Xu и соавт. [30] изучали последствия такого модулирующего риск развития шизофрении пренатального стрессора, как недостаточное питание в период беременности, для экспрессии генов в префронтальной коре и гиппокампе мышей и нашли, что в результате пренатальной нехватки пищи происходит перепрограммирование постнатальной экспрессии генов. В префронтальной коре отмечали изменение экспрессии генов, вовлеченных в нейротрансмиссию и обоняние, а в гиппокампе — связанных с синаптической передачей. При этом в гиппокампе имело место изменение профиля метилирования, преимущественно (в 87% случаев) заключавшееся в гиперметилировании ДНК.
J. Xu и соавт. [30] изучали последствия такого модулирующего риск развития шизофрении пренатального стрессора, как недостаточное питание в период беременности, для экспрессии генов в префронтальной коре и гиппокампе мышей и нашли, что в результате пренатальной нехватки пищи происходит перепрограммирование постнатальной экспрессии генов. В префронтальной коре отмечали изменение экспрессии генов, вовлеченных в нейротрансмиссию и обоняние, а в гиппокампе — связанных с синаптической передачей. При этом в гиппокампе имело место изменение профиля метилирования, преимущественно (в 87% случаев) заключавшееся в гиперметилировании ДНК.
Данные об отдельных генах-кандидатах шизофрении также указывают на возможную роль эпигенетических модификаций в развитии и изменчивости когнитивных нарушений у больных. Более того, эпигенетическими изменениями отчасти объясняется высокая вариативность результатов исследований ассоциаций. Например, однонуклеотидная замена (SNP от англ. Singlenucleotidepolymorphism) в локусе rs6265 (полиморфизм Val66Met) в гене BDNF, который рассматривается как модификатор ряда проявлений шизофрении, является так называемым CpG-полиморфизмом внутри CpG-динуклеотида (CpG-SNP): аллель Val содержит CpG, аллель Met — нет.

Одним из когнитивных эндофенотипов шизофрении является нарушение распознавания эмоциональной мимики. L. Rubin и соавт. [34] изучили связь метилирования участка, расположенного вблизи кодона гена окситоцина, с распознаванием эмоций и параметрами мозга у больных c эндогенными психозами (шизофрения и биполярные аффективные расстройства) и здоровых. Было обнаружено, что как у здоровых, так и страдающих шизофренией женщин уровни метилирования коррелируют с распознаванием эмоций и объемом мозга в префронтальных и височно-лимбических отделах. При этом все женщины характеризовались более высокими уровнями метилирования относительно мужчин и все больные шизофренией — относительно биполярных больных.
Было обнаружено, что как у здоровых, так и страдающих шизофренией женщин уровни метилирования коррелируют с распознаванием эмоций и объемом мозга в префронтальных и височно-лимбических отделах. При этом все женщины характеризовались более высокими уровнями метилирования относительно мужчин и все больные шизофренией — относительно биполярных больных.
Данные эпигенетики уже стали важным компонентом в комплексе данных, на основании которых исследователи пытаются восстановить путь от гена-кандидата к нарушениям когнитивной деятельности и от них к шизофрении. Примером может служить модель, использованная в работе I. Negrо́n-Oyarzo и соавт. [35], в которой дефицит регуляторно-исполнительских функций, являющийся важной характеристикой больных шизофренией, объясняется эпигенетическими изменениями гена RELN под действием пренатального стресса. RELN является одним из традиционных кандидатов для шизофрении и сопутствующих ей нарушений рабочей памяти. Он кодирует рилин — белок внеклеточного матрикса, вовлеченный в кортикогенез в эмбриональном периоде и в синаптическую пластичность, имеющую место после рождения. В ряде работ было выявлено снижение экспрессии RELN и гиперметилирование CpG-островка в его промоторе в префронтальной коре больных шизофренией. При этом на модельных животных продемонстрирована роль пренатального стресса в гиперметилировании промотора RELN в префронтальной коре. Основываясь на этих данных и учитывая место патологии префронтальной коры в патогенезе шизофрении и роль пренатального стресса в аномалиях развития этого региона мозга, I. Negrón-Oyarzo и соавт. [35] высказали предположение, что пренатальный стресс вызывает гиперметилирование в промоторе RELN, которое ведет к снижению его пренатальной транскрипции. Это влечет за собой сокращение числа интернейронов, синтезирующих ГАМК, нарушение размещения интернейронов в корковых слоях и редукцию длины дендритов пирамидных нейронов в префронтальной коре. Таким образом, пренатальный стресс нарушает эмбриональное развитие префронтальной коры, что проявляется аномалиями организации нервных сетей в этом регионе мозга.
В ряде работ было выявлено снижение экспрессии RELN и гиперметилирование CpG-островка в его промоторе в префронтальной коре больных шизофренией. При этом на модельных животных продемонстрирована роль пренатального стресса в гиперметилировании промотора RELN в префронтальной коре. Основываясь на этих данных и учитывая место патологии префронтальной коры в патогенезе шизофрении и роль пренатального стресса в аномалиях развития этого региона мозга, I. Negrón-Oyarzo и соавт. [35] высказали предположение, что пренатальный стресс вызывает гиперметилирование в промоторе RELN, которое ведет к снижению его пренатальной транскрипции. Это влечет за собой сокращение числа интернейронов, синтезирующих ГАМК, нарушение размещения интернейронов в корковых слоях и редукцию длины дендритов пирамидных нейронов в префронтальной коре. Таким образом, пренатальный стресс нарушает эмбриональное развитие префронтальной коры, что проявляется аномалиями организации нервных сетей в этом регионе мозга. На уровне поведения патология работы префронтальных отделов выражается в нарушениях регуляторно-исполнительских функций и повышает риск развития шизофрении.
На уровне поведения патология работы префронтальных отделов выражается в нарушениях регуляторно-исполнительских функций и повышает риск развития шизофрении.
Говоря об эпигенетике когнитивного дефицита при шизофрении, важно отметить, что изучение эпигенетических процессов должно пролить свет на механизмы не только нарушений развития нервной системы, ведущие к аномалиям когнитивных процессов, но и на механизмы резистентности когнитивных функций к действию патологических факторов, лежащих в основе возникновения и течения психоза. Известно, что когнитивная дисфункция при шизофрении характеризуется высокой вариативностью как выраженности, так и динамики после манифестации заболевания [36, 37]. При этом существует группа больных, общий интеллект которых остается стабильно высоким в ходе болезни [36]. Резистентность когнитивных процессов к развитию ведущей к психическому расстройству церебральной патологии нередко объясняют возможностью привлечения дополнительных нейрональных ресурсов, которые получили название мозговых или когнитивных резервов [38]. Их существование связывают с высокой пластичностью мозга, обусловленной количеством нейронов и/или мощностью существующих и возможностью образования новых нейронных сетей. Полагают, что когнитивные резервы формируются в процессе генотип-средового взаимодействия под влиянием обогащенной среды [39]. Показано [40], что физическая и умственная активность улучшает память у человека и животных и ведет к возрастанию когнитивных резервов и снижению риска потери памяти при нейродегенеративных заболеваниях. Описаны [41, 42] некоторые эпигенетические процессы, опосредующие влияние обогащенной среды на когнитивные функции. В частности, установлена роль ацетилирования гистонов в улучшении памяти в обогащенной среде как на ранних, так и поздних стадиях онтогенеза. Показано в том числе [43], что ацетилирование гистонов тесно связано с транскрипцией промотора гена BDNF — важнейшего молекулярного посредника действия обогащенной среды на синаптическую пластичность.
Их существование связывают с высокой пластичностью мозга, обусловленной количеством нейронов и/или мощностью существующих и возможностью образования новых нейронных сетей. Полагают, что когнитивные резервы формируются в процессе генотип-средового взаимодействия под влиянием обогащенной среды [39]. Показано [40], что физическая и умственная активность улучшает память у человека и животных и ведет к возрастанию когнитивных резервов и снижению риска потери памяти при нейродегенеративных заболеваниях. Описаны [41, 42] некоторые эпигенетические процессы, опосредующие влияние обогащенной среды на когнитивные функции. В частности, установлена роль ацетилирования гистонов в улучшении памяти в обогащенной среде как на ранних, так и поздних стадиях онтогенеза. Показано в том числе [43], что ацетилирование гистонов тесно связано с транскрипцией промотора гена BDNF — важнейшего молекулярного посредника действия обогащенной среды на синаптическую пластичность.
У больных шизофренией интегральным, хотя и непрямым индикатором когнитивных резервов может быть уровень образования или преморбидный интеллект [36, 38]. В последнее время накапливаются данные о роли эпигенетических механизмов в развитии общего интеллекта человека [27, 44, 45]. Так, C. Yu и соавт. [44] исследовали 17 пар монозиготных близнецов, у которых имелась большая внутрипарная разница в IQ — не менее одного стандартного отклонения, или 15 баллов (обычно она составляет около 6 баллов). Изучив метилирование 25 500 промоторов в этих дискордантных по интеллекту парах, авторы предложили 27 генов, несовпадение в метилировании которых может влиять на дискордантность по интеллекту, и выявили умеренную положительную корреляцию между различиями в метилировании и интеллекте. Полученные данные, однако, оказались не очень убедительными и противоречивыми, поскольку в этих работах использовались различные методы. В частности, между близнецами с высоким и невысоким интеллектом не было выявлено различий в транскрипции генов. K. Lillycrop и соавт. [27] в небольшой выборке нашли ассоциацию между пренатальным метилированием ряда участков генома (при использовании ДНК, выделенной из пуповины) с общим интеллектом ребенка, оцененным в 4-летнем возрасте.
В последнее время накапливаются данные о роли эпигенетических механизмов в развитии общего интеллекта человека [27, 44, 45]. Так, C. Yu и соавт. [44] исследовали 17 пар монозиготных близнецов, у которых имелась большая внутрипарная разница в IQ — не менее одного стандартного отклонения, или 15 баллов (обычно она составляет около 6 баллов). Изучив метилирование 25 500 промоторов в этих дискордантных по интеллекту парах, авторы предложили 27 генов, несовпадение в метилировании которых может влиять на дискордантность по интеллекту, и выявили умеренную положительную корреляцию между различиями в метилировании и интеллекте. Полученные данные, однако, оказались не очень убедительными и противоречивыми, поскольку в этих работах использовались различные методы. В частности, между близнецами с высоким и невысоким интеллектом не было выявлено различий в транскрипции генов. K. Lillycrop и соавт. [27] в небольшой выборке нашли ассоциацию между пренатальным метилированием ряда участков генома (при использовании ДНК, выделенной из пуповины) с общим интеллектом ребенка, оцененным в 4-летнем возрасте. Эти участки были связаны как с генами, которые ранее рассматривались в качестве кандидатов для когниций (например, гены, кодирующие транскрипционный фактор TCF4, антагонист рецептора интерлейкина-1 (IL1RN), ядерный эритроидный фактор (NFE2L2)), так и новыми. Причем многие из них кодируют транскрипционные факторы, вовлеченные в развитие диэнцефалона (ETS1, HES1, TCF4, NR4A2). Дальнейшее изучение одного из них — HES1 — на расширенных до 150—200 человек выборках подтвердило, что более высокий уровень перинатального метилирования некоторых участков его промотора ассоциирован с более высоким IQ в 4 года и лучшим выполнением тестов на регуляторно-исполнительские функции в 7 лет. Изменение в уровне метилирования HES1 на одно стандартное отклонение было связано с изменением IQ на 3,2 балла, в то время как самый большой эффект генетического полиморфизма (в гене транскрипционного фактора HMGA2 [46]) на интеллект составил 1,29 балла. A. Córdova-Palomera и соавт. [45] показали, что на внутрипарные различия монозиготных близнецов по общему интеллекту и рабочей памяти может влиять генетический полиморфизм гена ДНК-метилтрансферазы 3 (DNMT3B), кодирующего один из осуществляющих метилирование ДНК ферментов.
Эти участки были связаны как с генами, которые ранее рассматривались в качестве кандидатов для когниций (например, гены, кодирующие транскрипционный фактор TCF4, антагонист рецептора интерлейкина-1 (IL1RN), ядерный эритроидный фактор (NFE2L2)), так и новыми. Причем многие из них кодируют транскрипционные факторы, вовлеченные в развитие диэнцефалона (ETS1, HES1, TCF4, NR4A2). Дальнейшее изучение одного из них — HES1 — на расширенных до 150—200 человек выборках подтвердило, что более высокий уровень перинатального метилирования некоторых участков его промотора ассоциирован с более высоким IQ в 4 года и лучшим выполнением тестов на регуляторно-исполнительские функции в 7 лет. Изменение в уровне метилирования HES1 на одно стандартное отклонение было связано с изменением IQ на 3,2 балла, в то время как самый большой эффект генетического полиморфизма (в гене транскрипционного фактора HMGA2 [46]) на интеллект составил 1,29 балла. A. Córdova-Palomera и соавт. [45] показали, что на внутрипарные различия монозиготных близнецов по общему интеллекту и рабочей памяти может влиять генетический полиморфизм гена ДНК-метилтрансферазы 3 (DNMT3B), кодирующего один из осуществляющих метилирование ДНК ферментов. Авторы интерпретируют результаты в пользу роли этого фактора эпигенетических модификаций как посредника в отклике интеллекта на обогащенную или обедненную среду.
Авторы интерпретируют результаты в пользу роли этого фактора эпигенетических модификаций как посредника в отклике интеллекта на обогащенную или обедненную среду.
Подобные данные могут быть использованы для изучения и понимания механизмов сохранения высокого уровня когнитивного функционирования у некоторых больных шизофренией. Однако пока опубликована лишь одна работа [47], в которой авторы попытались непосредственно оценить связь между эпигенетическими маркерами и уровнем когнитивного дефицита больных. Был изучен статус метилирования 19 генов в аутопсийных образцах мозга 29 больных шизофренией пожилого возраста, который сравнили с премортальным когнитивным дефицитом. Изменений в уровне метилирования у больных ни с относительно сохранной, ни с нарушенной когнитивной функцией найдено не было. Этот негативный результат, однако, следует рассматривать с осторожностью, поскольку в работе оценивали грубые когнитивные нарушения (с помощью Mini Mental State Examination и опросников), которые не являются типичными для шизофрении.
Исследования влияния обогащенной среды на когнитивные функции важны для понимания механизмов действия когнитивно-поведенческой терапии и нейрокогнитивных тренингов больных шизофренией. Кроме того, эпигенетические модификации представляют собой обратимые процессы, в связи с чем являются привлекательными мишенями медикаментозного лечения [48, 49]. В модельных опытах на животных было установлено, что фармакологическое воздействие ведет к восстановлению активности генома и нормализации поведения, в том числе тех изменений, которые связаны с особенностями метилирования генов дофаминергического пути, вызванных стрессом или мутациями [50, 51]. Так, K. Brami-Cherrier и соавт. [51] продемонстрировали, что у мышей с мутацией, ответственной за отсутствие D2-ауторецепторов в мезенцефальных дофаминергических нейронах, изменяется геномный ландшафт в нейронах, получающих дофаминергические проекции и расположенных в разных регионах коры. В частности, в префронтальной коре наблюдалось снижение экспрессии 1930 генов и повышение — 2 генов. Существенная часть из них оказалась вовлечена в регуляцию транскрипции, включая моделирование хроматина. Мутантные мыши при этом демонстрировали изменения поведения, которые считаются характерными для психозов, в том числе нарушения рабочей памяти. Лечение в течение 2 нед агонистом рецепторов D2 квинпиролом вело предположительно через стимуляцию постсинаптических D2-рецепторов к восстановлению экспрессии генов в нейронах префронтальной коры и повышенных у мутантных мышей уровней h4K9me3 — индикатора метилирования гистонов до значений, наблюдаемых у животных без мутации.
Существенная часть из них оказалась вовлечена в регуляцию транскрипции, включая моделирование хроматина. Мутантные мыши при этом демонстрировали изменения поведения, которые считаются характерными для психозов, в том числе нарушения рабочей памяти. Лечение в течение 2 нед агонистом рецепторов D2 квинпиролом вело предположительно через стимуляцию постсинаптических D2-рецепторов к восстановлению экспрессии генов в нейронах префронтальной коры и повышенных у мутантных мышей уровней h4K9me3 — индикатора метилирования гистонов до значений, наблюдаемых у животных без мутации.
Приведенные результаты дают основание надеяться, что эпигенетические процессы послужат новой платформой для развития терапевтических подходов к коррекции когнитивных нарушений при шизофрении [52]. В качестве потенциальных средств в эпигенетической фармакологии рассматривают ингибиторы деацетилаз гистонов (HDAC) — ферментов, катализирующих удаление ацетильной группы с гистоновых N-концов и тем самым связанных с репрессией генов.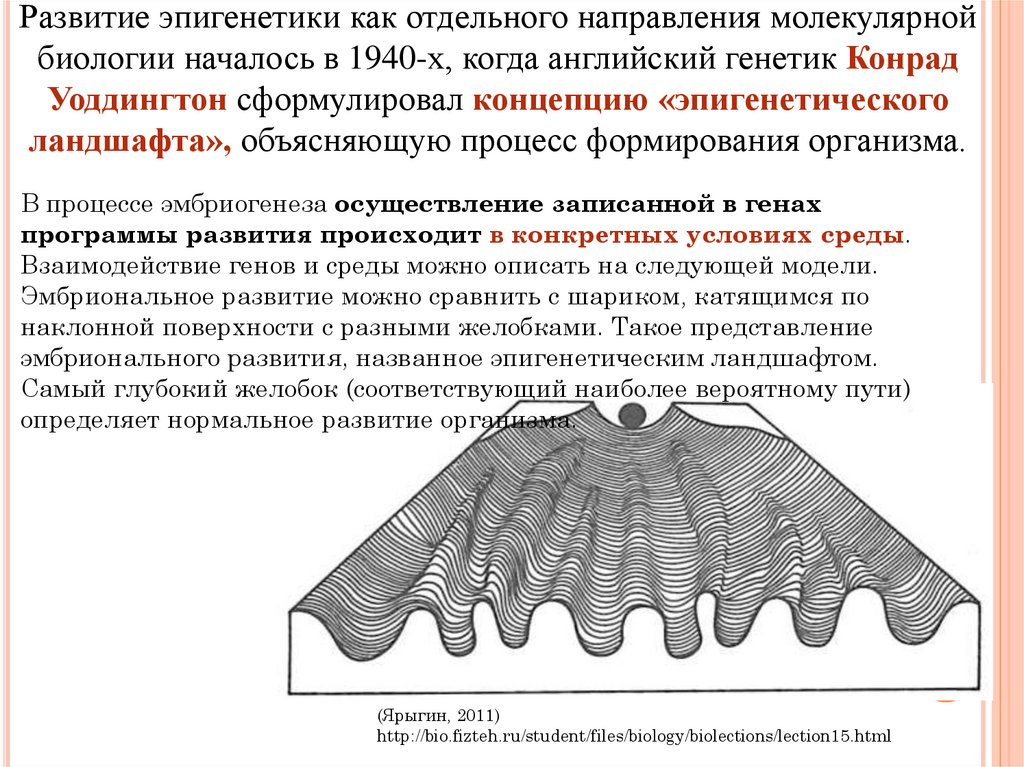 Кроме того, внимание привлечено к ингибиторам ферментов, катализирующих метилирование ДНК, — ДНК-метилтрансферазам (DNMT), а также веществам, способствующим деметилированию ДНК. Некоторые из этих препаратов, уже использующиеся в лечении рака и сердечной аритмии, представляются перспективными и в отношении воздействия на симптомы шизофрении. Например, ингибиторы DNMT снижают ферментативную активность DNMT и гиперметилирование ДНК в промоторах генов RELN и GAD1, тем самым нормализуя ГАМКергическую нейротрансмиссию [53]. Ряд авторов [54, 55] указывают, что ингибиторы деацетилаз гистонов могут быть использованы для коррекции когнитивной сферы больных шизофренией. Следует отметить, что в механизмы действия уже существующих антипсихотических препаратов вовлечены эпигенетические процессы [49, 56, 57]. Галоперидол связан с рядом модификаций, включая ацетилирование, фосфорилирование и фосфоацетилирование гистонов и метилирование ДНК[57]. Клозапин и сульпирид связаны с ацетилированием гистонов и метилированием ДНК [57].
Кроме того, внимание привлечено к ингибиторам ферментов, катализирующих метилирование ДНК, — ДНК-метилтрансферазам (DNMT), а также веществам, способствующим деметилированию ДНК. Некоторые из этих препаратов, уже использующиеся в лечении рака и сердечной аритмии, представляются перспективными и в отношении воздействия на симптомы шизофрении. Например, ингибиторы DNMT снижают ферментативную активность DNMT и гиперметилирование ДНК в промоторах генов RELN и GAD1, тем самым нормализуя ГАМКергическую нейротрансмиссию [53]. Ряд авторов [54, 55] указывают, что ингибиторы деацетилаз гистонов могут быть использованы для коррекции когнитивной сферы больных шизофренией. Следует отметить, что в механизмы действия уже существующих антипсихотических препаратов вовлечены эпигенетические процессы [49, 56, 57]. Галоперидол связан с рядом модификаций, включая ацетилирование, фосфорилирование и фосфоацетилирование гистонов и метилирование ДНК[57]. Клозапин и сульпирид связаны с ацетилированием гистонов и метилированием ДНК [57]. Эти препараты, в частности, способствуют деметилированию гена RELN in vivo; а при назначении вместе с этими препаратами вальпроата (ингибитор деацетилазы гистонов) их способность к деметилированию гена RELN усиливается [56]. При этом вальпроат и клозапин улучшают когнитивные функции у животных, на которых моделируют сходные с шизофренией нарушения [53]. Перспективным подходом является целевое изменение эпигенетического статуса конкретных терапевтических мишеней с помощью гибридных систем на основе метода редактирования генома CRISPR/CAS9 [58].
Эти препараты, в частности, способствуют деметилированию гена RELN in vivo; а при назначении вместе с этими препаратами вальпроата (ингибитор деацетилазы гистонов) их способность к деметилированию гена RELN усиливается [56]. При этом вальпроат и клозапин улучшают когнитивные функции у животных, на которых моделируют сходные с шизофренией нарушения [53]. Перспективным подходом является целевое изменение эпигенетического статуса конкретных терапевтических мишеней с помощью гибридных систем на основе метода редактирования генома CRISPR/CAS9 [58].
За 15 лет с начала молекулярно-генетических исследований когнитивных нарушений при шизофрении были достигнуты большие успехи в развитии молекулярно-генетических технологий и накоплены данные о генетических коррелятах связанных с заболеванием когнитивных нарушений. Однако до установления генетических основ нейрокогнитивного дефицита, которые могли бы пролить свет на его биологические механизмы и сделать возможным проведение персонифицированного обследования больных на этапе манифестации первого приступа психоза с целью подбора соответствующей терапии, предстоит еще много работы.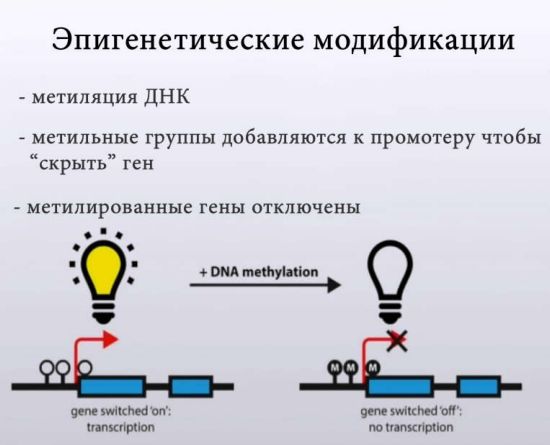 Важную роль в достижении поставленных целей должна сыграть эпигенетика. Можно ожидать, что для такого сложного мультифакторного признака, как когнитивный дефицит, эпигенетические маркеры станут более точными предикторами, чем генетические. Хотя эпигенетика и эпигеномная медицина представляют собой новые отрасли науки о поведении и пока лишь приблизились к изучению когнитивного дефицита при шизофрении, в их рамках уже получен ряд интересных данных, проливающих свет на механизмы генотип-средового взаимодействия, определяющие развитие и функционирование мозга, которые лежат в основе когниций. Фармакоэпигенетика должна способствовать поиску путей устранения, казалось бы, безвозвратно утерянных когнитивных, поведенческих, сенсомоторных функций посредством эпигенетического репрограммирования судьбы стволовых клеток, способствуя восстановлению целостности нейронных сетей, пластичности и связности [52]. Однако когнитивный дефицит представляет собой результат сложного взаимодействия патологических и компенсаторных процессов, происходящих в мозге под действием как генетических, так и средовых факторов.
Важную роль в достижении поставленных целей должна сыграть эпигенетика. Можно ожидать, что для такого сложного мультифакторного признака, как когнитивный дефицит, эпигенетические маркеры станут более точными предикторами, чем генетические. Хотя эпигенетика и эпигеномная медицина представляют собой новые отрасли науки о поведении и пока лишь приблизились к изучению когнитивного дефицита при шизофрении, в их рамках уже получен ряд интересных данных, проливающих свет на механизмы генотип-средового взаимодействия, определяющие развитие и функционирование мозга, которые лежат в основе когниций. Фармакоэпигенетика должна способствовать поиску путей устранения, казалось бы, безвозвратно утерянных когнитивных, поведенческих, сенсомоторных функций посредством эпигенетического репрограммирования судьбы стволовых клеток, способствуя восстановлению целостности нейронных сетей, пластичности и связности [52]. Однако когнитивный дефицит представляет собой результат сложного взаимодействия патологических и компенсаторных процессов, происходящих в мозге под действием как генетических, так и средовых факторов.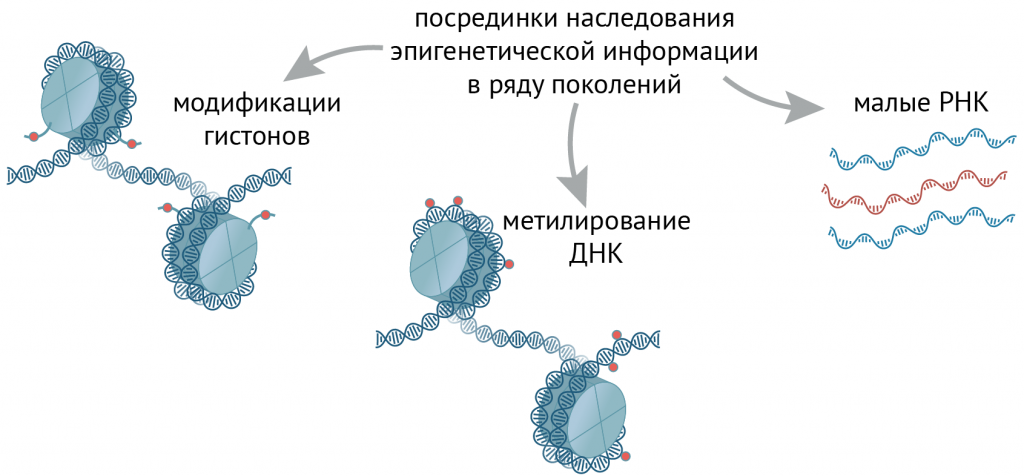 Поэтому следует ожидать, что вариативность эпигенетических меток окажется весьма высокой, а следовательно, воспроизводимость результатов и размеры эффектов отдельных маркеров — еще более низкими, чем при исследовании структурных изменений последовательности ДНК. Возможно, эта проблема будет решена в комплексных исследованиях геномной организации отдельных индивидов. На это как на основную перспективу развития генетических подходов в психиатрии указывают I. Houston и соавт. [59]. Как отмечают эти авторы, следует ожидать все более широкого применения картирования эпигенетических меток с высоким разрешением в аутопсийном мозге и культурах нейрональных клеток, происходящих из плюрипотентных клеток, вместе с транскрипторным профилированием и полногеномным секвенированием для выявления молекулярной патологии конкретных случаев с диагнозами шизофрении, аутизма и других психиатрических расстройств.
Поэтому следует ожидать, что вариативность эпигенетических меток окажется весьма высокой, а следовательно, воспроизводимость результатов и размеры эффектов отдельных маркеров — еще более низкими, чем при исследовании структурных изменений последовательности ДНК. Возможно, эта проблема будет решена в комплексных исследованиях геномной организации отдельных индивидов. На это как на основную перспективу развития генетических подходов в психиатрии указывают I. Houston и соавт. [59]. Как отмечают эти авторы, следует ожидать все более широкого применения картирования эпигенетических меток с высоким разрешением в аутопсийном мозге и культурах нейрональных клеток, происходящих из плюрипотентных клеток, вместе с транскрипторным профилированием и полногеномным секвенированием для выявления молекулярной патологии конкретных случаев с диагнозами шизофрении, аутизма и других психиатрических расстройств.
Исследование выполнено за счет гранта Российского научного фонда (проект № 16−15−00056).
Конфликт интересов отсутствует.
1Этот вопрос рассматривался нами в предыдущей публикации: см. Журнал неврологии и психиатрии им. С.С. Корсакова, 2016, 116.
Эпигенетика как доказательная база влияния образа жизни на здоровье и болезни
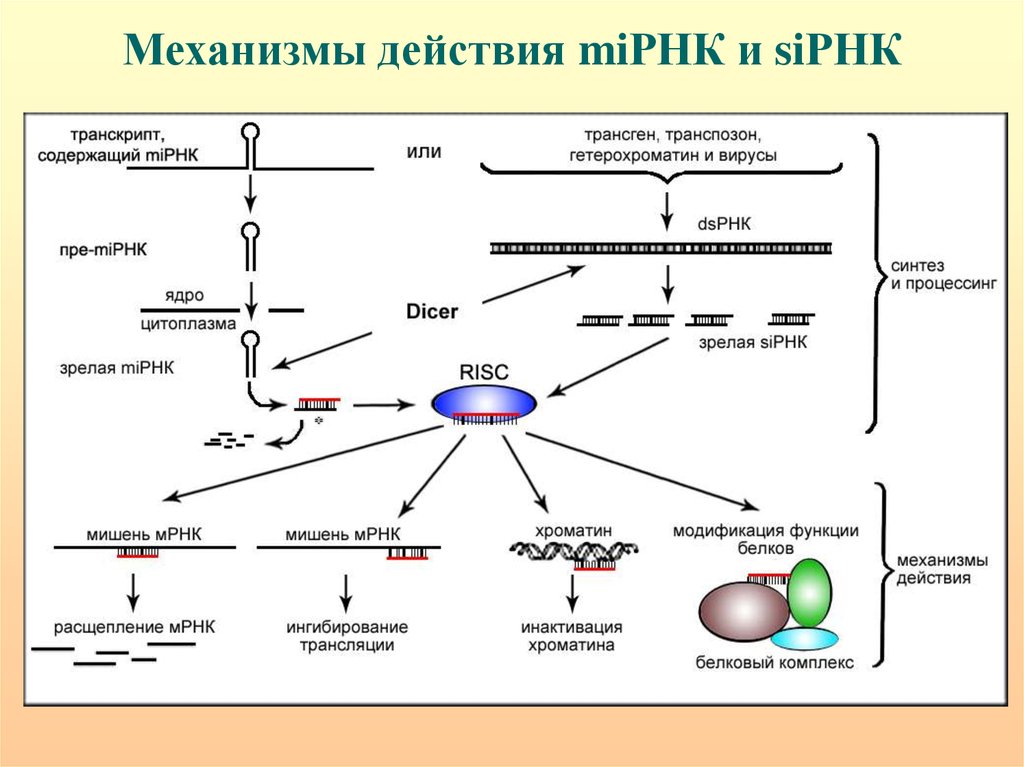
Эпигенетика — новое, интенсивно развивающееся направление генетики. Эпигенетика представляет собой науку о наследуемых свойствах организма, которые не связаны с изменением собственно нуклеотидной последовательности ДНК и могут быть не прямо, а опосредованно закодированы в геноме. К числу известных эпигенетических механизмов (сигналы) относятся энзиматическое метилирование ДНК, гистоновый код (разные энзиматические модификации гистонов — ацетилирование, метилирование, фосфорилирование, убиквитинирование и т. д.) и «замалчивание» генов малыми РНК (miRNA, siRNA) [1]. Метилирование ДНК является одним из самых популярных эпигенетических механизмов, который осуществляется ДНК-метилтрансферазой по основанию цитозин в последовательности нуклеотидов CpG (5’-C-фосфат-G-3’), где С — цитозин, G — гуанидин. Около 70% промоторов человеческих генов характеризуется высоким содержанием последовательности CpG. Метилирование подавляет экспрессию генов, а деметилирование, наоборот, индуцируя ацетилирование гистонов и связанные с этим изменения от гетерохроматина к эухроматину, активирует транскрипцию генов. Накопление стресса усваивается как приобретенная информация, обозначаемая термином «эпигенетическая память», и может передаваться по наследству. С профилактической точки зрения крайне важным является раскрытие эпигенетических механизмов влияния образа жизни и условий существования организма на здоровье.
Около 70% промоторов человеческих генов характеризуется высоким содержанием последовательности CpG. Метилирование подавляет экспрессию генов, а деметилирование, наоборот, индуцируя ацетилирование гистонов и связанные с этим изменения от гетерохроматина к эухроматину, активирует транскрипцию генов. Накопление стресса усваивается как приобретенная информация, обозначаемая термином «эпигенетическая память», и может передаваться по наследству. С профилактической точки зрения крайне важным является раскрытие эпигенетических механизмов влияния образа жизни и условий существования организма на здоровье.
Цель исследования — провести анализ эпигенетических исследований влияния образа жизни на здоровье и болезни.
Материал и методы
Аналитическое исследование публикаций, представленных в электронных библиотеках PubMed, ResearchGate, e-library, CiberLeninka и др.
Результаты
История развития эпигенетических подходов к доказательству влияния образа жизни на здоровье и болезни. Работы Дэвида Баркера в 80—90-х гг. XX века заложили основу программирования риска хронических заболеваний взрослых в критический период формирования организма [2]. Д. Баркер [3] показал, что неадекватное снабжение организма ребенка питательными веществами или кислородом формирует резистентность к инсулину [3]. По мнению D. Lawlor и соавт. [4], внутриутробные воздействия определяют риск развития ишемической болезни сердца (ИБС) у взрослых, который при обратной связи с массой тела при рождении опосредуется резистентностью к инсулину. Сегодня теория «Истоки развития здоровья и болезней» (Developmental Origins of Health and Disease, DOHaD) предполагает, что экспозиция в ранний период развития организма играет решающую роль в определении риска метаболических заболеваний у взрослых, что доказано в эпигенетических исследованиях в отношении метаболических нарушений, ожирения и хронических заболеваний [5—8]. Дисрегуляция miРНК вызывает изменения в структуре генов, контролирующих воспаление, липидный обмен, резистентность к инсулину и адипогенез [9].
Работы Дэвида Баркера в 80—90-х гг. XX века заложили основу программирования риска хронических заболеваний взрослых в критический период формирования организма [2]. Д. Баркер [3] показал, что неадекватное снабжение организма ребенка питательными веществами или кислородом формирует резистентность к инсулину [3]. По мнению D. Lawlor и соавт. [4], внутриутробные воздействия определяют риск развития ишемической болезни сердца (ИБС) у взрослых, который при обратной связи с массой тела при рождении опосредуется резистентностью к инсулину. Сегодня теория «Истоки развития здоровья и болезней» (Developmental Origins of Health and Disease, DOHaD) предполагает, что экспозиция в ранний период развития организма играет решающую роль в определении риска метаболических заболеваний у взрослых, что доказано в эпигенетических исследованиях в отношении метаболических нарушений, ожирения и хронических заболеваний [5—8]. Дисрегуляция miРНК вызывает изменения в структуре генов, контролирующих воспаление, липидный обмен, резистентность к инсулину и адипогенез [9]. Современные представления о детском ожирении как составной части метаболического синдрома базируются на многофакторности его происхождения и ключевой роли эпигенетики в передаче риска ожирения потомству за счет генетического наследования однонуклеотидных полиморфизмов в локусах адипокинов и их рецепторов и влияния микробиоты кишечника, участвующей в регуляции массы тела [10].
Современные представления о детском ожирении как составной части метаболического синдрома базируются на многофакторности его происхождения и ключевой роли эпигенетики в передаче риска ожирения потомству за счет генетического наследования однонуклеотидных полиморфизмов в локусах адипокинов и их рецепторов и влияния микробиоты кишечника, участвующей в регуляции массы тела [10].
Эпигенетические механизмы программирования состояния здоровья потомства, обусловленные питанием матери во время зачатия и беременности. Питание матери во время зачатия ребенка и в период его раннего развития может эпигенетическим путем инициировать метаболические сдвиги у потомства, известные как «программирование питанием». Драматический рост распространенности аллергических заболеваний связывают с пищевым программированием специфически уязвимой в раннем возрасте иммунной системы. Глубокое понимание эпигенетики и других биологических процессов в раннем возрасте может привести к разработке диетических стратегий, обеспечивающих более устойчивое состояние именной системы в ранний период и снижающих бремя многих воспалительных заболеваний, а не только аллергии [11].
В опытах на животных показано, что сниженная калорийность питания матери эпигенетически индуцирует усиление возрастной непереносимости глюкозы у поросят [12]; добавка бетаина беременным свиньям увеличивает содержание холестерина в печени неонатальных поросят посредством эпигенетических правил метаболических генов холестерина [13]. Долгосрочное потребление высоких доз никотинамида (витамина РР) самками крыс, в том числе при беременности, может быть фактором риска метаболических аномалий у потомства, связанных с метилированием генов и инсулинорезистентностью, а фолиевой кислоты — увеличивает опухолевый генез молочной железы, но снижает риск колоректального рака и ряда врожденных дефектов сердца у потомства [14—16]. Введение 150 мг фолиевой кислоты в яйца улучшает рост бройлеров и укрепляет взаимосвязь между иммунной функцией и эпигенетической регуляцией иммунных генов путем изменения конформации хроматина и метилирования промотеров гистонов [17]. У людей эффект воздействия фолиевой кислоты на эпигенетическую регуляцию фосфоенолпируваткарбоксикиназы — ключевого фермента в образовании глюкозы из пировиноградной кислоты и гомеостаза глюкозы зависит от периода жизненного цикла и пола [18]. Материнский статус фолата, регулируемый диетическими и генетическими факторами на ранних стадиях беременности, предположительно может влиять на риск расстройств аутистического спектра у людей, однако данные об эпигенетическом воздействии пока ограниченны [19].
Материнский статус фолата, регулируемый диетическими и генетическими факторами на ранних стадиях беременности, предположительно может влиять на риск расстройств аутистического спектра у людей, однако данные об эпигенетическом воздействии пока ограниченны [19].
Таким образом, сердечно-сосудистая патология, ожирение, аутоиммунные проявления, сахарный диабет (СД) и предположительно аутизм эпигенетически связаны с образом жизни матери, пренатальным и постнатальным периодами, относимыми к критическим в отношении здоровья в будущем, и могут регулироваться такими пищевыми компонентами, как фолаты и фолиевая кислота.
Эпигенетические механизмы старения. Старение как совокупность изменений, постепенно увеличивающих вероятность смерти, с эпигенетической точки зрения характеризуется воспроизводимым в независимых выборках гипометилированием CpG-последовательностей [20]. Возрастные изменения эпигенетических меток могут приводить к снижению иммунной функции, что способствует увеличению заболеваемости пожилых людей, для поддержания здоровья которых здоровый образ жизни (ЗОЖ) на протяжении всей жизни, учитывая пожизненную эпигеномную регуляцию во врожденных иммунных клетках, в лимфоцитах Т- и В- под действием внутренних и внешних факторов, может быть самым эффективным способом профилактики заболеваний [21, 22].
Естественные возрастные изменения, приводящие к высококонкурентной экспрессии генов с явными последствиями для клеточной дифференциации и риском начала заболевания, играют несомненную роль в формировании болезней сердечно-сосудистой системы. Модифицируемые и немодифицируемые факторы риска эпигенетически изменяют экспрессию генов в возрасте, ускоряя эпигенетические «часы», прежде избавлявшие человека от сердечно-сосудистых заболеваний. Ускоренное сосудистое старение и, как следствие, снижение возрастного порога заболевания, обусловленное эпигенетическим возрастом, не совпадающим с хронологическим, вызывает серьезную озабоченность. Вместе с тем адекватное питание и физическая активность оказывают синергическое воздействие на здоровье сердечно-сосудистой системы, представляя собой мощную потенциальную эпигенетическую точку вмешательства с целью коррекции управленческих стратегий в отношении сердечно-сосудистой системы, направленных на «хорошее старение» [23]. Детальное изучение дисрегулированных эпигенетических механизмов, связанных с СД и его сосудистыми осложнениями (кардиомиопатия, нефропатия, ретинопатия, синдром диабетической стопы), может раскрыть столь необходимые новые лекарственные мишени для профилактики сосудистых заболеваний в целом [24, 25].
Одним из примеров профилактики возрастных изменений может служить длительный прием фолиевой кислоты пожилыми здоровыми людьми, вызвавший глобальное метилирование ДНК, причем, несмотря на препозицию связи нейродеструктивных процессов с аберрантным метилированием ДНК в лейкоцитах, когнитивные способности пожилых только улучшились [26]. Однако не только внешние воздействия, но и генетические факторы влияют на эпигеномные изменения. Так, в лонгитюдном исследовании психических расстройств у пожилых жителей Австралии (n=1863) показано, что метилирование и модификация связи между депрессией и метилированием ДНК находятся под влиянием генетических вариантов ангиотензинпревращающего фермента, играющего ключевую роль в регуляции гипоталамо-гипофизарно-надпочечниковой системы [27].
Из 500 тыс. локусов, метилирование которых связано с риском смерти по причине рака, отобраны 10 участков CpG, строго коррелирующих с риском смерти [28]. Метилирование ДНК клеток крови по локусам AHRR, 6p21. 33 и F2RL3 является прогностическим для развития рака легких и может быть использовано для идентификации групп риска при скрининге [29]. Идентифицированные локусы mQTLs, оказывающие влияние на метилирование участков CpG, имеют особое значение при их использовании в качестве маркеров метилирования ДНК в связанных с курением сравнительных популяционных исследованиях. H. Brenner [30] отмечает: «Неблагоприятный статус метилирования может измениться после прекращения курения, и риск смертности может значительно снизиться… Профилактика или вмешательство в состояния, связанные с курением (ДНК-метилирование), могут эффективно способствовать предупреждению преждевременной смерти, учитывая обратимость индуцированных курением метиломных аберраций». В эпигенетических исследованиях пациентов с колоректальным раком (n=1836) выявлены специфические изменения miРНК, связанные с опухолевыми проявлениями и косвенно определяющие выживаемость пациентов [31].
33 и F2RL3 является прогностическим для развития рака легких и может быть использовано для идентификации групп риска при скрининге [29]. Идентифицированные локусы mQTLs, оказывающие влияние на метилирование участков CpG, имеют особое значение при их использовании в качестве маркеров метилирования ДНК в связанных с курением сравнительных популяционных исследованиях. H. Brenner [30] отмечает: «Неблагоприятный статус метилирования может измениться после прекращения курения, и риск смертности может значительно снизиться… Профилактика или вмешательство в состояния, связанные с курением (ДНК-метилирование), могут эффективно способствовать предупреждению преждевременной смерти, учитывая обратимость индуцированных курением метиломных аберраций». В эпигенетических исследованиях пациентов с колоректальным раком (n=1836) выявлены специфические изменения miРНК, связанные с опухолевыми проявлениями и косвенно определяющие выживаемость пациентов [31].
Таким образом, возрастные изменения связаны с активацией генов гипометилированием и иными эпигенетическими и генетическими модуляциями, что обусловливает снижение активности иммунной системы, сердечно-сосудистые болезни, СД и его сосудистые осложнения, депрессию и рак. Интервенции посредством изменения образа жизни могут снизить риск смерти и продлить здоровую жизнь пожилых людей благодаря эпигенетическим механизмам, обеспечивая популярную концепцию «хорошего старения». «Связь между диетой и эпигенетическими изменениями, с одной стороны, и между эпигенетическими изменениями и раком — с другой, подтверждается как обсервационными исследованиями на людях, так и опытами на животных. Однако вывод о том, что диета напрямую связана с эпигенетическими изменениями и что эти эпигенетические изменения непосредственно увеличивают или уменьшают риск развития рака человека, гораздо менее определен» [32].
Интервенции посредством изменения образа жизни могут снизить риск смерти и продлить здоровую жизнь пожилых людей благодаря эпигенетическим механизмам, обеспечивая популярную концепцию «хорошего старения». «Связь между диетой и эпигенетическими изменениями, с одной стороны, и между эпигенетическими изменениями и раком — с другой, подтверждается как обсервационными исследованиями на людях, так и опытами на животных. Однако вывод о том, что диета напрямую связана с эпигенетическими изменениями и что эти эпигенетические изменения непосредственно увеличивают или уменьшают риск развития рака человека, гораздо менее определен» [32].
Выявленные к 2011 г. эпигенетические механизмы, обеспечивающие влияние элементов образа жизни и среды на здоровье. К 2011 г. стало известно, что потребление фолатов, эпигаллокатехин-3-галлатов зеленого чая, селена, а также физическая активность, табакокурение, материнская диета и табакокурение матери во время беременности, вредное потребление алкоголя, воздействие поллютантов окружающей среды (мышьяк, хром, аэрозоли, бензол, полициклическихе ароматические углеводороды и стойкие органические соединения), а также старение, психологический стресс и сменная работа оказывают влияние на экспрессию генов путем изменения метилирования ДНК. Потребление полифенольных соединений и селена с пищей, а также физическая активность ведут к ковалентной модификации (ацетилированию) гистоновых белков. Физическая активность, курение сигарет и внутриутробные условия, в частности связанные с курением табака матерью во время гестации, регулируется экспрессией miРНК путем метилирования ДНК в miРНК локусах [33].
Потребление полифенольных соединений и селена с пищей, а также физическая активность ведут к ковалентной модификации (ацетилированию) гистоновых белков. Физическая активность, курение сигарет и внутриутробные условия, в частности связанные с курением табака матерью во время гестации, регулируется экспрессией miРНК путем метилирования ДНК в miРНК локусах [33].
Эпигенетические механизмы воздействия питания и нутриентов на современном этапе. За прошедшие 7 лет многие эпигенетические механизмы были уточнены и расширены, в том числе в отношении питания и приема нутриентов. Так, показано, что куркумин изменяет эпигенетические маркеры, подавляя активацию ядерного транскрипционного фактора каппа-би В-клеток (NF-κB), тем самым уменьшая воспалительные реакции. Эпигаллокатехин гидрат также может снижать риск воспаления, сердечной травмы и окислительного повреждения, вызванного поллютантами окружающей среды, посредством эпигенетической регуляции генов провоспалительных мишеней NF-κB [34]. Обладающие антиоксидантной активностью полифенолы, содержащие катехины, подавляют активность ферментов и эпигенетически активируют «молчащие» гены. Некоторые нутриенты, включая фолиевую кислоту, кобаламин, рибофлавин, пиридоксин и метионин, играют решающую роль в метаболизме 1-углерода, непосредственно воздействуя на S-аденозил-L-метионин. Соевые полифенолы блокируют ДНК-метилтрансферазы и гистондеацетилазы, обеспечивая обратное аберрантное метилирование локусов CpG. Сульфорафан, обнаруженный в брокколи, нормализует метилирование ДНК и активирует экспрессию miR-140, которая в свою очередь подавляет SOX9 и ALDh2 и уменьшает рост опухолей [35]. В четырех европейских когортах (n=3096) только среди потребителей чая, но не кофе, женского пола выявлено два дифференциально метилированных CpG-сайта в составе генов DNAJC16 и TTC17, участвующих в опухолевых процессах и метаболизме эстрогенов [36]. Токоферолы — класс химических соединений, представляющих собой метилированные фенолы, многие из которых объединены названием «витамин E», — изменяя профили miРНК у пациентов, инфицированных вирусом гепатита B, проявляют антивирусную активность [37].
Обладающие антиоксидантной активностью полифенолы, содержащие катехины, подавляют активность ферментов и эпигенетически активируют «молчащие» гены. Некоторые нутриенты, включая фолиевую кислоту, кобаламин, рибофлавин, пиридоксин и метионин, играют решающую роль в метаболизме 1-углерода, непосредственно воздействуя на S-аденозил-L-метионин. Соевые полифенолы блокируют ДНК-метилтрансферазы и гистондеацетилазы, обеспечивая обратное аберрантное метилирование локусов CpG. Сульфорафан, обнаруженный в брокколи, нормализует метилирование ДНК и активирует экспрессию miR-140, которая в свою очередь подавляет SOX9 и ALDh2 и уменьшает рост опухолей [35]. В четырех европейских когортах (n=3096) только среди потребителей чая, но не кофе, женского пола выявлено два дифференциально метилированных CpG-сайта в составе генов DNAJC16 и TTC17, участвующих в опухолевых процессах и метаболизме эстрогенов [36]. Токоферолы — класс химических соединений, представляющих собой метилированные фенолы, многие из которых объединены названием «витамин E», — изменяя профили miРНК у пациентов, инфицированных вирусом гепатита B, проявляют антивирусную активность [37].
Эпигенетические механизмы воздействия ЗОЖ как интегрального показателя на здоровье. ЗОЖ в совокупности его элементов также может играть определенную роль в регуляции метилирования ДНК. Так, выявлены высокие уровни индекса глобального метилирования ДНК и гена TNF-α противовоспалительного цитокина в белых клетках крови в группе здоровых молодых людей (n=156) со средней нормальной массой тела, соблюдающих ЗОЖ, против контрольной группы с метаболическими нарушениями. Среди элементов ЗОЖ 1-й группы выявлены потребление рационального количества энергии и микроэлементов с пищей, большее число занимающихся спортом лиц и меньшее число курящих [38]. С другой стороны, пациенты с СД 1-го типа (Чили) без осложнений относительно контрольной группы без СД показали достоверно более высокий уровень метилирования промотора гена TNF-α [39].
Функциональная значимость эпигенетических механизмов как интерфейса между модификациями образа жизни и фенотипическими изменениями подчеркивается обширным перепрограммированием эпигенома диетой и физическими упражнениями.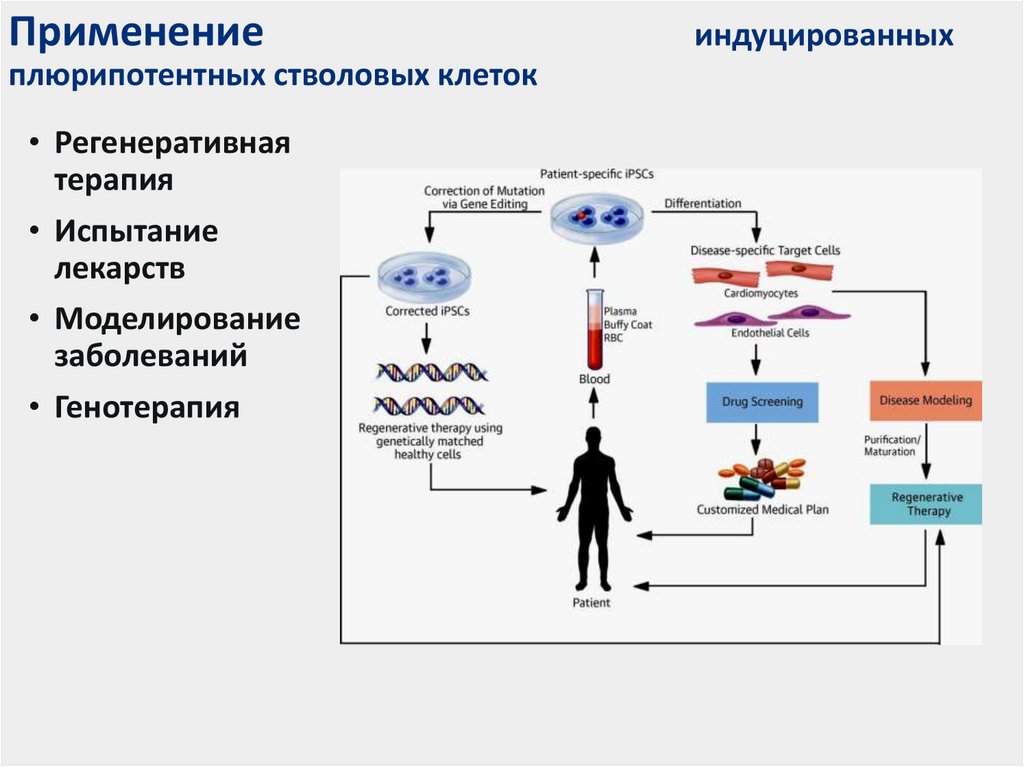 У мышей физические нагрузки и двигательная активность благодаря их защитным эффектам на фоне питания фастфудом, для которого характерно высокое содержание жиров, сахара и соли, предотвращали вызванное фастфудом гиперметилирование ДНК в клетках печени, особенно у промоторов и энхансеров. Вместе с тем ослабление гипометилирования в определенных участках ДНК наблюдалось только частично. Без физической нагрузки наблюдалось тотальное гиперметилирование и значительное увеличение специфических для печени энхансеров, что предполагает частичную потерю идентичности гепатоцитов, а гиперметилирование множества генных промоторов было связано с ингибированием развития ткани и промотированием канцерогенных процессов [40]. Коррелирующие со снижением массы тела изменения в эпигенетических модификациях 12 геномных локусов, из которых два расположены вблизи ассоциированных с потерей массы тела генов RUNX3 и NAMPT, выявлены у женщин 55—70 лет без инвалидности (n=20) на протяжении 6 мес, участвовавших в программе группового санитарного просвещения по вопросам значимости отказа от сидячего образа жизни для здоровья (9 занятий по 2 ч), против контрольной группы (6 занятий иной тематики по 1 ч).
У мышей физические нагрузки и двигательная активность благодаря их защитным эффектам на фоне питания фастфудом, для которого характерно высокое содержание жиров, сахара и соли, предотвращали вызванное фастфудом гиперметилирование ДНК в клетках печени, особенно у промоторов и энхансеров. Вместе с тем ослабление гипометилирования в определенных участках ДНК наблюдалось только частично. Без физической нагрузки наблюдалось тотальное гиперметилирование и значительное увеличение специфических для печени энхансеров, что предполагает частичную потерю идентичности гепатоцитов, а гиперметилирование множества генных промоторов было связано с ингибированием развития ткани и промотированием канцерогенных процессов [40]. Коррелирующие со снижением массы тела изменения в эпигенетических модификациях 12 геномных локусов, из которых два расположены вблизи ассоциированных с потерей массы тела генов RUNX3 и NAMPT, выявлены у женщин 55—70 лет без инвалидности (n=20) на протяжении 6 мес, участвовавших в программе группового санитарного просвещения по вопросам значимости отказа от сидячего образа жизни для здоровья (9 занятий по 2 ч), против контрольной группы (6 занятий иной тематики по 1 ч). В виде проекта авторы предложили потенциальный эпигенетический предиктор снижения массы тела на основе базового метилирования ДНК в 5 CpG-сайтах [41].
В виде проекта авторы предложили потенциальный эпигенетический предиктор снижения массы тела на основе базового метилирования ДНК в 5 CpG-сайтах [41].
Эпигенетические механизмы повреждающего воздействия экспозиции мелкодисперсным аэрозолем. Эпигенетические подтверждения вредного воздействия найдены в отношении вдыхания мелкодисперсных аэрозолей (взвешенные частицы), которые ВОЗ в 2005 г. отнесла к стохастическим факторам, не имеющим порога воздействия. Показано, что экспонирование аэрозолем с размером частиц не более 10 мкм (РМ10) повышает активность гистонацетилтрансферазы, катализирующей модификацию гистонов, и уровень ацетилированного гистона типа Н4 и таким образом способствует высвобождению воспалительных цитокинов [42]. Школьники (n=900) с воспалением дыхательных путей даже при кратковременном вдыхании самой мелкодисперсной пыли с размером частиц до 2,5 мкм (PM2,5) и наиболее опасной ввиду легкого проникновения сквозь биологические барьеры фракции аэрозолей проявили большую генетическую и эпигенетическую восприимчивость к этому типу аэрозолей [43]. РМ2,5 при 2-часовой экспозиции в концентрации 250 мг/м3 индуцирует метилирование в генах, участвующих в метаболизме митохондриальной энергии реакций окисления, снижаемое под действием витаминов группы В в составе аэрозоля, что позволяет использовать данный метод для индивидуальной профилактики вредного воздействия аэрозолей в промышленных районах с частыми пиками выбросов PM2,5 [44].
РМ2,5 при 2-часовой экспозиции в концентрации 250 мг/м3 индуцирует метилирование в генах, участвующих в метаболизме митохондриальной энергии реакций окисления, снижаемое под действием витаминов группы В в составе аэрозоля, что позволяет использовать данный метод для индивидуальной профилактики вредного воздействия аэрозолей в промышленных районах с частыми пиками выбросов PM2,5 [44].
Заключение
Изменения, происходящие в живом организме на всех этапах от зачатия и внутриутробного развития до старости и обусловленные воздействием элементов образа жизни и факторов окружающей среды, опосредованы регуляцией активности генов эпигенетическими механизмами, включающими метилирование ДНК, модификацию гистонов и «замалчивание» генов, промотеров или энхансеров малыми РНК. Многоуровневая система эпигенетических механизмов регулирует (изменяет) экспрессию определенных генов, задействованных в формировании положительных или отрицательных для здоровья ответов организма, в зависимости от специфических характеристик внешних факторов и механизмов их воздействия. Сегодня некоторые аспекты эпигенетической регуляции раскрыты. Например, установлено, что при старении экспрессия генов возрастает за счет инверсии метилирования ДНК в сторону гипометилирования, в том числе с вовлечением модификации гистонов, что и обусловливает формирование бремени неинфекционных хронических болезней в старости, многие из которых могут быть эпигенетически детерминированы в раннем возрасте образом жизни матери и состоянием ее здоровья при зачатии и беременности или даже получены по наследству. Дисрегуляция малых РНК обусловливает изменения в структурах генов, контролирующих воспаление и метаболические нарушения, что также обусловливает формирование неинфекционных болезней. Однако эпигенетические механизмы воздействия многих факторов не до конца выяснены и адекватно встроены в систему эпигенетического регулирования человеческого организма или еще не исследованы.
Сегодня некоторые аспекты эпигенетической регуляции раскрыты. Например, установлено, что при старении экспрессия генов возрастает за счет инверсии метилирования ДНК в сторону гипометилирования, в том числе с вовлечением модификации гистонов, что и обусловливает формирование бремени неинфекционных хронических болезней в старости, многие из которых могут быть эпигенетически детерминированы в раннем возрасте образом жизни матери и состоянием ее здоровья при зачатии и беременности или даже получены по наследству. Дисрегуляция малых РНК обусловливает изменения в структурах генов, контролирующих воспаление и метаболические нарушения, что также обусловливает формирование неинфекционных болезней. Однако эпигенетические механизмы воздействия многих факторов не до конца выяснены и адекватно встроены в систему эпигенетического регулирования человеческого организма или еще не исследованы.
В настоящее время становится очевидным, что, несмотря на необходимость уточнения многих эпигенетических механизмов воздействия внешних и поведенческих факторов, эпигенетика обеспечивает профилактическую медицину и гигиену не только информацией о возможных эпигенетических точках вмешательства ЗОЖ в широком понимании этого термина, но и молекулярной доказательной базой профилактических мероприятий. Возможность эффективного управления здоровьем через эпигенетические механизмы в любой период жизни человека отражено в концепции развития здоровья на протяжении всего жизненного цикла (Life Сourse Health Development — LCHD), разработанной под руководством Neal Halfon, в соответствии с которой здоровье представляет собой динамический процесс, начинающийся до зачатия и продолжающийся на протяжении всей жизни, и должно развиваться (укрепляться) во всех периодах жизненного цикла человека, поскольку негативные изменения в состоянии здоровья обратимы, их можно скорректировать даже в старости. Хотя основные процессы развития человека генетически запрограммированы, экспрессия генов модифицируется предыдущей и текущей средой и поведением [45]. В 2014 г. N. Halfon и соавт. [46] ставят вопрос о необходимости реформы общественного здоровья на основе теории LCHD и вносят предложения по внедрению инноваций, которые могли бы ускорить перевод принципов развития здоровья в практику менеджмента здоровья на протяжении всей жизни, которые соответствуют прецизионной (персонифицированной) медицине.
Возможность эффективного управления здоровьем через эпигенетические механизмы в любой период жизни человека отражено в концепции развития здоровья на протяжении всего жизненного цикла (Life Сourse Health Development — LCHD), разработанной под руководством Neal Halfon, в соответствии с которой здоровье представляет собой динамический процесс, начинающийся до зачатия и продолжающийся на протяжении всей жизни, и должно развиваться (укрепляться) во всех периодах жизненного цикла человека, поскольку негативные изменения в состоянии здоровья обратимы, их можно скорректировать даже в старости. Хотя основные процессы развития человека генетически запрограммированы, экспрессия генов модифицируется предыдущей и текущей средой и поведением [45]. В 2014 г. N. Halfon и соавт. [46] ставят вопрос о необходимости реформы общественного здоровья на основе теории LCHD и вносят предложения по внедрению инноваций, которые могли бы ускорить перевод принципов развития здоровья в практику менеджмента здоровья на протяжении всей жизни, которые соответствуют прецизионной (персонифицированной) медицине. В руководстве «Handbook of Life Course Health Development» [47] обобщается и анализируется растущая база знаний о возможностях и перспективах развитии здоровья на протяжении всей жизни.
В руководстве «Handbook of Life Course Health Development» [47] обобщается и анализируется растущая база знаний о возможностях и перспективах развитии здоровья на протяжении всей жизни.
Автор заявляет об отсутствии конфликта интересов.
The authors declare no conflicts of interest.
Сведения об авторах
Максименко Людмила Витальевна, к.биол.н., доц. [Ludmila V. Maksimenko, PhD, Associate Professor]; адрес: 117198, Россия, Москва,ул. Миклухо-Маклая, 6 [address: 6, Miklucho-Maklaya St., 117198, Moscow, Russia]; https://orcid.org/0000-0003-4048-855X; eLibrary SPIN: 1344-7617; e-mail: [email protected]
Эпигенетический подход к посттравматическому стрессу: аспекты крысиных моделей
1. Градус Дж. Л., Цинь П., Линкольн А. К. Посттравматическое стрессовое расстройство и завершенный суицид. Am J Эпидемиол. 2010;171(06):721–727. [PubMed] [Google Scholar]
2. Форте Г., Фавьери Ф., Тамбелли Р. , Касагранде М. Пандемия COVID-19 среди населения Италии: проверка опросника посттравматического стрессового расстройства и распространенность симптоматики посттравматического стрессового расстройства. Общественное здравоохранение Int J Environ Res. 2020;17(11):4151–4164. [Бесплатная статья PMC] [PubMed] [Google Scholar]
, Касагранде М. Пандемия COVID-19 среди населения Италии: проверка опросника посттравматического стрессового расстройства и распространенность симптоматики посттравматического стрессового расстройства. Общественное здравоохранение Int J Environ Res. 2020;17(11):4151–4164. [Бесплатная статья PMC] [PubMed] [Google Scholar]
3. Hossack MR, Reid MW, Aden JK, Gibbons T, Noe JC, Willis AM. Неблагоприятный детский опыт, гены и риск посттравматического стрессового расстройства у солдат: исследование метилирования Mil Med 2020185(3-4):377–384 . [PubMed] [Google Scholar]
4. Золадз П. Р., Даймонд Д. М. Модель посттравматического стрессового расстройства у животных на основе психосоциального стресса на основе хищников: доклиническая оценка травматического стресса на когнитивном, гормональном, фармакологическом, сердечно-сосудистом и эпигенетическом уровнях анализа Exp Neurol 2016284( ч. Б): 211–219. [PubMed] [Академия Google]
5. Voisey J, Young RM, Lawford BR, Morris C P. Прогресс в понимании генетики посттравматического стрессового расстройства. J Тревожное расстройство. 2014;28(08):873–883. [PubMed] [Google Scholar]
J Тревожное расстройство. 2014;28(08):873–883. [PubMed] [Google Scholar]
6. Соломон З., Микулинсер М. Траектории посттравматического стрессового расстройства: 20-летнее продольное исследование. Am J Психиатрия. 2006;163(04):659–666. [PubMed] [Google Scholar]
7. Борганс Б., Хомберг Дж. Р. Животные модели посттравматического стрессового расстройства: обзор того, что используется в исследованиях. Всемирная психиатрия. 2015;5(04):387–396. [Бесплатная статья PMC] [PubMed] [Google Scholar]
8. Warren B L, Vialou V F, Iñiguez S D. Нейробиологические последствия наблюдения стрессовых событий у взрослых мышей. Биол психиатрия. 2013;73(01):7–14. [PMC free article] [PubMed] [Google Scholar]
9. Антельман С. М. Зависимая от времени сенсибилизация как краеугольный камень нового подхода к фармакотерапии – лекарства как чужеродные стрессовые раздражители. Наркотики Dev Res. 1988; 14:1–30. [Google Scholar]
10. Louvart H, Maccari S, Ducrocq F, Thomas P, Darnaudéry M. Долгосрочные поведенческие изменения у самок крыс после однократного сильного удара током по ноге с последующими ситуационными напоминаниями. Психонейроэндокринология. 2005;30(04):316–324. [PubMed] [Академия Google]
Долгосрочные поведенческие изменения у самок крыс после однократного сильного удара током по ноге с последующими ситуационными напоминаниями. Психонейроэндокринология. 2005;30(04):316–324. [PubMed] [Академия Google]
11. Павлик А.С., Джха С.К., Бреннан Ф.Х., Моррисон А.Р., Росс Р.Дж. Модель нарушений сна у грызунов при посттравматическом стрессовом расстройстве: роль контекста после формирования страха. Биол психиатрия. 2005;57(03):268–277. [PubMed] [Google Scholar]
12. Рау В., Фанселоу М. С. Воздействие стрессора вызывает длительное улучшение обучения страху у крыс. Стресс. 2009;12(02):125–133. [PubMed] [Google Scholar]
13. Либерзон И., Крстов М., Янг Э. А. Стресс-рестресс: влияние на АКТГ и быстрая обратная связь. Психонейроэндокринология. 1997;22(06):443–453. [PubMed] [Google Scholar]
14. Balkaya M, Prinz V, Custodis F. Стресс ухудшает функцию эндотелия и ишемический инсульт через глюкокортикоиды. Гладить. 2011;42(11):3258–3264. [PubMed] [Google Scholar]
15. Золадз П.Р., Конрад С.Д., Флешнер М., Даймонд Д.М. Острые эпизоды воздействия хищников в сочетании с хронической социальной нестабильностью как модель посттравматического стрессового расстройства на животных. Стресс. 2008;11(04):259–281. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Золадз П.Р., Конрад С.Д., Флешнер М., Даймонд Д.М. Острые эпизоды воздействия хищников в сочетании с хронической социальной нестабильностью как модель посттравматического стрессового расстройства на животных. Стресс. 2008;11(04):259–281. [Бесплатная статья PMC] [PubMed] [Google Scholar]
16. Ким Х.Г., Харрисон П.А., Годекер А.Л., Музыка С.Н. Посттравматическое стрессовое расстройство у женщин, получающих дородовую помощь в трех квалифицированных медицинских центрах федерального уровня. Health Matern Child Health J. 2014;18(05):1056–1065. [PubMed] [Google Scholar]
17. Золадз П. Р., Флешнер М., Даймонд Д. М. Психосоциальная животная модель посттравматического стрессового расстройства вызывает длительную травматическую память, усиление общей тревожности и сходные с посттравматическим стрессовым расстройством глюкокортикоидные аномалии. Психонейроэндокринология. 2012;37(09):1531–1545. [PubMed] [Академия Google]
18. Бланшар Р. Дж., Бланшар Д. С., Роджерс Дж., Вайс С. М. Характеристика и моделирование защитного поведения против хищников. Neurosci Biobehav Rev. 1990;14(04):463–472. [PubMed] [Google Scholar]
М. Характеристика и моделирование защитного поведения против хищников. Neurosci Biobehav Rev. 1990;14(04):463–472. [PubMed] [Google Scholar]
19. Апфельбах Р., Бланшар С. Д., Бланшар Р. Дж., Хейс Р. А., МакГрегор И. С. Влияние запахов хищников на виды млекопитающих-жертв: обзор полевых и лабораторных исследований. Neurosci Biobehav Rev. 2005;29(08):1123–1144. [PubMed] [Google Scholar]
20. Коэн Х., Гева А.Б., Матар М.А., Зохар Дж., Каплан З. Поведенческие реакции на посттравматический стресс у инбредных линий мышей: может ли генетическая предрасположенность объяснить фенотипическую уязвимость? Int J Neuropsychopharmacol. 2008;11(03):331–349.. [PubMed] [Google Scholar]
21. Careaga M BL, Girardi C EN,suchecki D. Понимание посттравматического стрессового расстройства через формирование, угасание и реконсолидацию страха. Neurosci Biobehav Rev. 2016; 71:48–57. [PubMed] [Google Scholar]
22. Zovkic I B, Sweatt J D. Эпигенетические механизмы выученного страха: последствия посттравматического стрессового расстройства. Нейропсихофармакология. 2013;38(01):77–93. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Нейропсихофармакология. 2013;38(01):77–93. [Бесплатная статья PMC] [PubMed] [Google Scholar]
23. Квапис Дж. Л., Вуд М. А. Эпигенетические механизмы формирования страха: последствия для лечения посттравматического стрессового расстройства. Тренды Нейроси. 2014;37(12):706–720. [Бесплатная статья PMC] [PubMed] [Google Scholar]
24. Сааведра-Родригес Л., Фейг Л. А. Хроническая социальная нестабильность вызывает тревогу и неполноценное социальное взаимодействие между поколениями. Биол психиатрия. 2013;73(01):44–53. [Бесплатная статья PMC] [PubMed] [Google Scholar]
25. Иманака А., Моринобу С., Токи С., Ямаваки С. Важность ранней среды в развитии поведения, подобного посттравматическому стрессовому расстройству. Поведение мозга Res. 2006;173(01):129–137. [PubMed] [Google Scholar]
26. Cloitre M, Stolbach BC, Herman J L. Подход к комплексному посттравматическому стрессовому расстройству с точки зрения развития: детская и взрослая кумулятивная травма как предикторы сложности симптомов. J Травматический стресс. 2009 г.;22(05):399–408. [PubMed] [Google Scholar]
J Травматический стресс. 2009 г.;22(05):399–408. [PubMed] [Google Scholar]
27. Пуллиам Дж. В., Давагрех А. М., Алема-Менсах Э., Плотски П. М. Стресс социального поражения вызывает длительные изменения акустического испуга и увеличения массы тела у самцов крыс Long Evans. J Psychiatr Res. 2010;44(02):106–111. [Бесплатная статья PMC] [PubMed] [Google Scholar]
28. Yang R, Daigle B J, Jr., Muhie SY. Основные модульные биомаркеры крови и мозга в мышиной модели социального поражения при посттравматическом стрессовом расстройстве. BMC Сист Биол. 2013;7:80. [Бесплатная статья PMC] [PubMed] [Google Scholar]
29. Урсано Р. Дж., Ли Х., Чжан Л. Модели посттравматического стресса и травматического стресса: важность исследований «от постели к скамейке и до кровати» Prog Brain Res. 2008; 167: 203–215. [PubMed] [Google Scholar]
30. Yehuda R, Hoge CW, McFarlane A C. Посттравматическое стрессовое расстройство. Праймеры Nat Rev Dis. 2015;1:15057. [PubMed] [Google Scholar]
31. Фрилингсдорф Х., Бат К.Г., Солиман Ф., Дифеде Дж., Кейси Б.Дж., Ли Ф.С. Вариант нейротрофического фактора головного мозга Val66Met эндофенотипы: значение для посттравматического стрессового расстройства. Энн Н.Ю. Академия наук. 2010;1208:150–157. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Фрилингсдорф Х., Бат К.Г., Солиман Ф., Дифеде Дж., Кейси Б.Дж., Ли Ф.С. Вариант нейротрофического фактора головного мозга Val66Met эндофенотипы: значение для посттравматического стрессового расстройства. Энн Н.Ю. Академия наук. 2010;1208:150–157. [Бесплатная статья PMC] [PubMed] [Google Scholar]
32. Matsuoka Y, Nishi D, Noguchi H, Kim Y, Hashimoto K. Продольные изменения нейротрофического фактора головного мозга в сыворотке крови у выживших после несчастных случаев с посттравматическим стрессовым расстройством. Нейропсихобиология. 2013;68(01):44–50. [PubMed] [Google Scholar]
33. Maddox S A, Watts C S, Doyère V, Schafe GE. Природный ингибитор гистон-ацетилтрансферазы, полученный из Garcinia indica, ослабляет недавно приобретенные и реактивированные воспоминания о страхе. ПЛОС Один. 2013;8(01):e54463. [Бесплатная статья PMC] [PubMed] [Google Scholar]
34. Sartor CE, McCutcheon VV, Pommer NE. Общий генетический и экологический вклад в посттравматическое стрессовое расстройство и алкогольную зависимость у молодых женщин. Психомед. 2012;41(07):1497–1505. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Психомед. 2012;41(07):1497–1505. [Бесплатная статья PMC] [PubMed] [Google Scholar]
35. Leen-Feldner EW, Feldner MT, Knapp A, Bunaciu L, Blumenthal H, Amstadter AB. Психологические и биологические корреляты родительского посттравматического стресса у потомства: обзор литература и исследовательская программа. Clin Psychol Rev. 2013;33(08):1106–1133. [PubMed] [Академия Google]
36. Lambert J E, Holzer J, Hasbun A. Связь между тяжестью посттравматического стрессового расстройства у родителей и психологическим дистрессом у детей: метаанализ. J Травматический стресс. 2014;27(01):9–17. [PubMed] [Google Scholar]
37. Басавараджаппа Б.С., Суббанна С. Регуляция метилирования гистонов при нейродегенеративных заболеваниях. Int J Mol Sci. 2021;22(09):4654-4681.. [Бесплатная статья PMC] [PubMed] [Google Scholar]
38. Lubin FD, Sweatt JD. Киназа IkappaB регулирует структуру хроматина во время реконсолидации условных воспоминаний о страхе. Нейрон. 2007;55(06):942–957. [PMC free article] [PubMed] [Google Scholar]
39. Мэддокс С.А., Шафе Г.Э. Эпигенетические изменения в латеральной миндалине необходимы для реконсолидации павловской памяти о страхе. Выучить Мем. 2011;18(09):579–593. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Мэддокс С.А., Шафе Г.Э. Эпигенетические изменения в латеральной миндалине необходимы для реконсолидации павловской памяти о страхе. Выучить Мем. 2011;18(09):579–593. [Бесплатная статья PMC] [PubMed] [Google Scholar]
40. Maddox SA, Watts CS, Schafe GE. Активность гистон-ацетилтрансферазы p300/CBP необходима для вновь приобретенных и реактивированных воспоминаний о страхе в латеральной миндалевидном теле. Выучить Мем. 2013;20(02):109–119. [Бесплатная статья PMC] [PubMed] [Google Scholar]
41. Stafford JM, Raybuck JD, Ryabinin AE, Lattal KM. Повышение ацетилирования гистонов в гиппокампово-инфралимбической сети усиливает угашение страха. Биол психиатрия. 2012;72(01):25–33. [PMC бесплатная статья] [PubMed] [Google Scholar]
42. Webb WM, Sanchez RG, Perez G. Динамическая ассоциация эпигенетических меток h4K4me3 и ДНК 5hmC в дорсальном гиппокампе и передней поясной коре после реактивации памяти о страхе Neurobiol Learn Мем 2017142 (часть А): 66–78. [PubMed] [Академия Google]
43. Takei S., Morinobu S., Yamamoto S., Fuchikami M., Matsumoto T., Yamawaki S. Усиленная передача сигналов BDNF/TrkB в гиппокампе в ответ на обусловливание страха в животной модели посттравматического стрессового расстройства. J Psychiatr Res. 2011;45(04):460–468. [PubMed] [Google Scholar]
Takei S., Morinobu S., Yamamoto S., Fuchikami M., Matsumoto T., Yamawaki S. Усиленная передача сигналов BDNF/TrkB в гиппокампе в ответ на обусловливание страха в животной модели посттравматического стрессового расстройства. J Psychiatr Res. 2011;45(04):460–468. [PubMed] [Google Scholar]
44. Lee J B, Wei J, Liu W, Cheng J, Feng J, Yan Z. Гистоновая деацетилаза 6 блокирует синаптическое действие острого стресса в префронтальной коре. Дж. Физиол. 2012;590(07):1535–1546. [Бесплатная статья PMC] [PubMed] [Google Scholar]
45. Блуин А.М., Силливан С.Е., Джозеф Н.Ф., Миллер С.А. Потенциал эпигенетики в моделях обучения посттравматическому стрессу, усиленному стрессом. Выучить Мем. 2016;23(10):576–586. [Бесплатная статья PMC] [PubMed] [Google Scholar]
46. Jin B, Li Y, Robertson K D. Метилирование ДНК: высшее или подчиненное в эпигенетической иерархии? Гены Рак. 2011;2(06):607–617. [Бесплатная статья PMC] [PubMed] [Google Scholar]
47. Khare T, Pai S, Koncevicius K. 5-hmC в головном мозге широко распространен в синаптических генах и показывает различия на границе экзон-интрон. Nat Struct Mol Biol. 2012;19(10): 1037–1043. [Бесплатная статья PMC] [PubMed] [Google Scholar]
5-hmC в головном мозге широко распространен в синаптических генах и показывает различия на границе экзон-интрон. Nat Struct Mol Biol. 2012;19(10): 1037–1043. [Бесплатная статья PMC] [PubMed] [Google Scholar]
48. Мехта Д., Брюниг Д., Каррильо-Роа Т. Анализ метилирования ДНК всего генома у ветеранов боевых действий выявил новый локус посттравматического стрессового расстройства. Acta Psychiatr Scand. 2017;136(05):493–505. [PubMed] [Google Scholar]
49. Лог М. В., Амштадтер А. Б., Бейкер Д. Г. Консорциум психиатрической геномики, рабочая группа по посттравматическому стрессовому расстройству: посттравматическое стрессовое расстройство вступает в эпоху крупномасштабного геномного сотрудничества. Нейропсихофармакология. 2015;40(10):2287–2297. [Бесплатная статья PMC] [PubMed] [Google Scholar]
50. Алмли Л.М., Стивенс Дж.С., Смит А.К. Полногеномный идентифицированный вариант риска посттравматического стрессового расстройства представляет собой локус количественного признака метилирования и придает сниженную корковую активацию страху. лица. Am J Med Genet B Neuropsychiatr Genet. 2015; 8: 327–336. [Бесплатная статья PMC] [PubMed] [Google Scholar]
лица. Am J Med Genet B Neuropsychiatr Genet. 2015; 8: 327–336. [Бесплатная статья PMC] [PubMed] [Google Scholar]
51. Xie P, Kranzler HR, Yang C, Zhao H, Farrer L A, Gelernter J. Полногеномное ассоциативное исследование выявляет новые локусы предрасположенности к посттравматическому стрессовому расстройству. Биол психиатрия. 2013;74(09): 656–663. [Статья PMC бесплатно] [PubMed] [Google Scholar]
52. Duncan L E, Ratanathharathorn A, Aiello AE. Самый большой GWAS посттравматического стрессового расстройства (N = 20 070) дает генетическое совпадение с шизофренией и половыми различиями в наследственности. Мол Психиатрия. 2018;23(03):666–673. [Бесплатная статья PMC] [PubMed] [Google Scholar]
53. Рабочая группа Центра исследований, образования и клинического центра Среднеатлантических психических заболеваний по делам ветеранов . Эшли-Кох А.Е., Гарретт М.Е., Гибсон Дж. Общегеномное ассоциативное исследование посттравматического стрессового расстройства у когорты ветеранов эпохи Ирака и Афганистана. J Аффективное расстройство. 2015;184(184):225–234. [Бесплатная статья PMC] [PubMed] [Google Scholar]
J Аффективное расстройство. 2015;184(184):225–234. [Бесплатная статья PMC] [PubMed] [Google Scholar]
54. Kilaru V, Iyer S V, Almli LM. Анализ генов на основе всего генома предполагает связь между нейролигином 1 (NLGN1) и посттравматическим стрессовым расстройством. Трансл Психиатрия. 2016;6(05):e820. [Бесплатная статья PMC] [PubMed] [Google Scholar]
55. Программа совместных исследований Департамента по делам ветеранов (№ 575B) и программа Million Veteran Program. Гелернтер Дж., Сан Н., Полиманти Р. Общегеномное ассоциативное исследование посттравматического стрессового расстройства, повторно переживающего симптомы у> 165 000 ветеранов США. Нат Нейроски. 2019;22(09):1394–1401. [Бесплатная статья PMC] [PubMed] [Google Scholar]
56. Rutten B PF, Vermetten E, Vinkers CH. Продольный анализ метилома ДНК военнослужащих, находящихся на вооружении, позволяет выявить локусы предрасположенности к посттравматическому стрессовому расстройству. Мол Психиатрия. 2018;23(05):1145–1156. [Бесплатная статья PMC] [PubMed] [Google Scholar]
[Бесплатная статья PMC] [PubMed] [Google Scholar]
57. Hammamieh R, Chakraborty N, Gautam A. Статус метилирования полногеномной ДНК, связанный с клиническими показателями посттравматического стрессового расстройства у ветеранов OIF / OEF. Трансл Психиатрия. 2017;7(07):e1169. [Бесплатная статья PMC] [PubMed] [Google Scholar]
58. van Dongen J, Slagboom PE, Draisma H HM, Martin NG, Boomsma DI. Неизменная ценность исследований близнецов в эпоху омики. Нат Рев Жене. 2012;13(09):640–653. [PubMed] [Google Scholar]
59. Zhang T Y, Labonté B, Wen X L, Turecki G, Meaney M J. Эпигенетические механизмы ранней регуляции окружающей средой экспрессии генов глюкокортикоидных рецепторов гиппокампа у грызунов и людей. Нейропсихофармакология. 2013;38(01):111–123. [Бесплатная статья PMC] [PubMed] [Google Scholar]
60. Klengel T, Mehta D, Anacker C. Аллель-специфическое деметилирование ДНК FKBP5 опосредует взаимодействие генов и детских травм. Нат Нейроски. 2013;16(01):33–41. [Бесплатная статья PMC] [PubMed] [Google Scholar]
[Бесплатная статья PMC] [PubMed] [Google Scholar]
61. Li X, Wei W, Zhao Q Y. Накопление 5-гидроксиметилцитозина, опосредованное Tet3, в неокортексе способствует быстрой поведенческой адаптации. Proc Natl Acad Sci U S A. 2014;111(19):7120–7125. [Бесплатная статья PMC] [PubMed] [Google Scholar]
62. Ким Г.С., Смит А.К., Нивергельт С.М., Уддин М. Нейроэпигенетика посттравматического стрессового расстройства. Prog Mol Biol Transl Sci. 2018; 158: 227–253. [Бесплатная статья PMC] [PubMed] [Google Scholar]
63. Roth TL, Zoladz PR, Sweatt JD, Diamond DM. Эпигенетическая модификация ДНК Bdnf гиппокампа у взрослых крыс в модели посттравматического стрессового расстройства на животных. J Psychiatr Res. 2011;45(07):919–926. [Бесплатная статья PMC] [PubMed] [Google Scholar]
64. Гиллеспи С. Ф., Брэдли Б., Мерсер К. Травматическое воздействие и расстройства, связанные со стрессом, у пациентов первичной медико-санитарной помощи в центре города. Генерал Хосп Психиатрия. 2009;31(06):505–514. [Бесплатная статья PMC] [PubMed] [Google Scholar]
2009;31(06):505–514. [Бесплатная статья PMC] [PubMed] [Google Scholar]
65. de Quervain D J, Aerni A, Schelling G, Roozendaal B. Глюкокортикоиды и регуляция памяти в норме и при болезни. Передний нейроэндокринол. 2009 г.;30(03):358–370. [PubMed] [Google Scholar]
66. Иегуда Р., Даскалакис Н.П., Десарно Ф. Эпигенетические биомаркеры как предикторы и корреляты улучшения симптомов после психотерапии у ветеранов боевых действий с посттравматическим стрессовым расстройством. Фронтовая психиатрия. 2013;4:118. [Бесплатная статья PMC] [PubMed] [Google Scholar]
67. Collins LJ, Schönfeld B, Chen X S. Сан-Диего, Калифорния: Academic Press; 2011. Эпигенетика некодирующих РНК; стр. 49–61. [Google Scholar]
68. Куреши И.А., Мелер М.Ф. Новые роли некодирующих РНК в эволюции, развитии, пластичности и заболеваниях мозга. Нат Рев Нейроски. 2012;13(08):528–541. [Бесплатная статья PMC] [PubMed] [Google Scholar]
69. Даскалакис Н.П., Риджал С.М., Кинг С., Хакинс Л.М.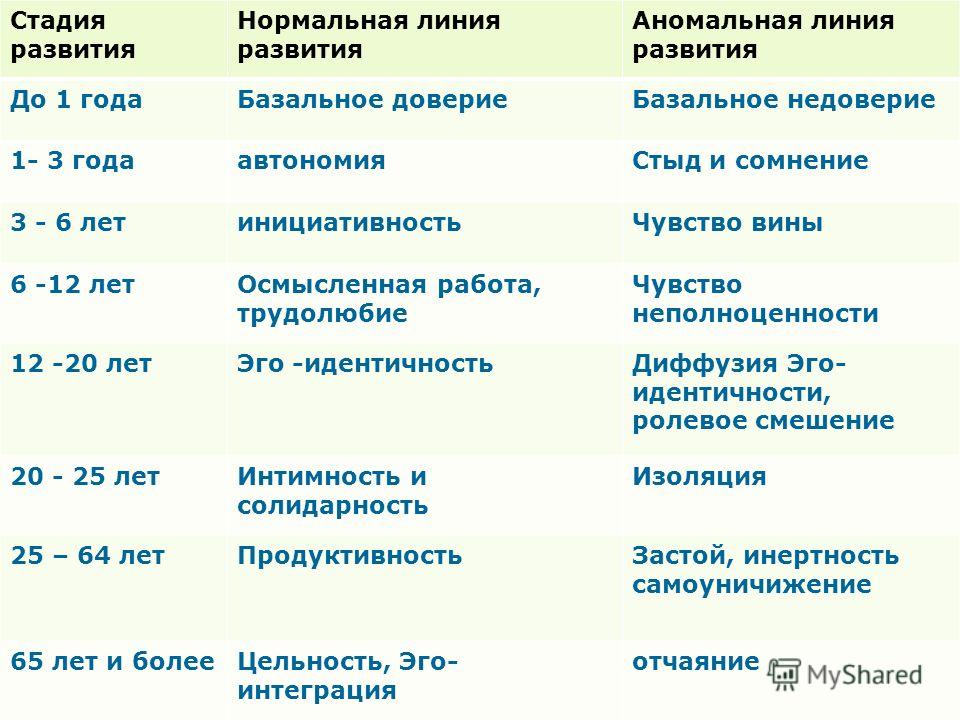 , Ресслер К.Дж. Современные генетические и эпигенетические подходы к посттравматическому стрессу. Curr Psychiatry Rep. 2018; 20 (05): 30. [PMC free article] [PubMed] [Google Scholar]
, Ресслер К.Дж. Современные генетические и эпигенетические подходы к посттравматическому стрессу. Curr Psychiatry Rep. 2018; 20 (05): 30. [PMC free article] [PubMed] [Google Scholar]
70. Амбескович М., Бабенко О., Ильницкий Ю., Ковальчук И., Колб Б., Мец Г. А.С. Наследственный стресс изменяет траектории психического здоровья на протяжении всей жизни и корковую нейроморфологию посредством эпигенетической регуляции. Научный доклад 2019; 9 (01): 6389. [Бесплатная статья PMC] [PubMed] [Google Scholar]
71. Maurel O M, Torrisi S A, Barbagallo C. Нарушение регуляции miR-15a-5p, miR-497a-5p и миР-511-5p связаны с модуляцией BDNF и FKBP5 в областях мозга восприимчивых и устойчивых мышей, связанных с ПТСР. Int J Mol Sci. 2021;22(10):5157–5177. [PMC free article] [PubMed] [Google Scholar]
72. Li C, Liu Y, Liu D, Jiang H, Pan F. Динамические изменения экспрессии miR-34c в гипоталамусе самцов крыс после травматического стресса в раннем подростковом возрасте. Нейр Пласт. 2016;2016:5.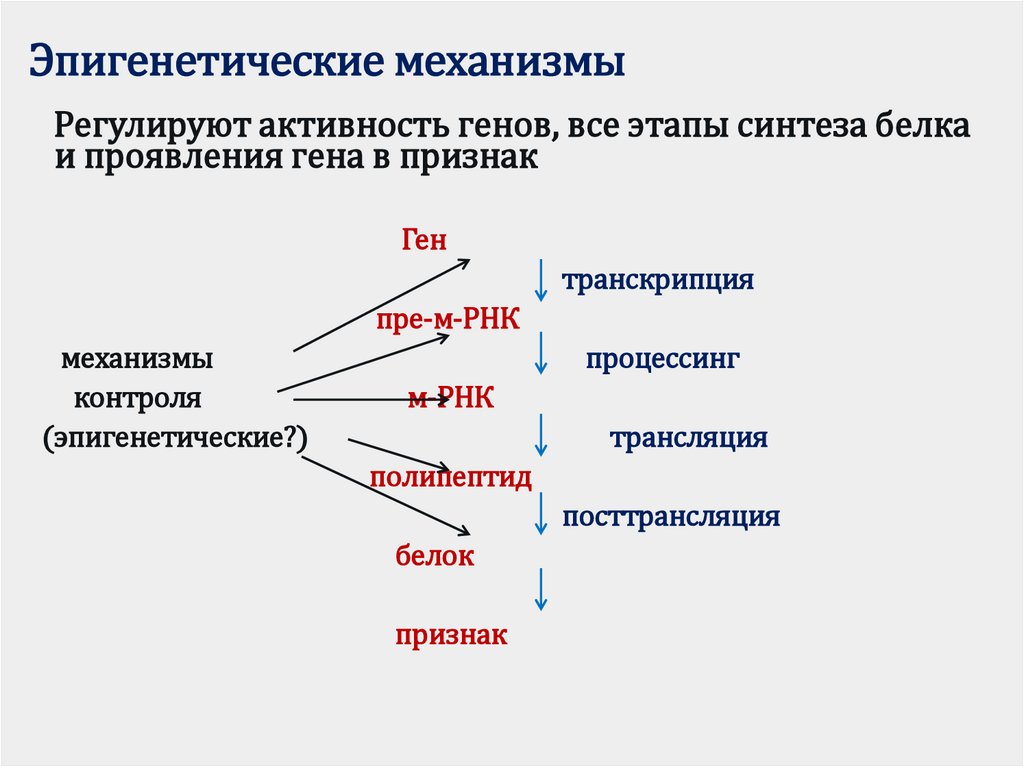 249893E6. [Бесплатная статья PMC] [PubMed] [Google Scholar]
249893E6. [Бесплатная статья PMC] [PubMed] [Google Scholar]
73. Kang J I, Kim TY, Choi J H, So HS, Kim S J. Аллель-специфический уровень метилирования ДНК FKBP5 связан с посттравматическим стрессовым расстройством. Психонейроэндокринология. 2019;103:1–7. [PubMed] [Google Scholar]
Эпигенетические подходы к психическим расстройствам
1. Henikoff S, Matzke MA. Изучение и объяснение эпигенетических эффектов. Тенденции Жене. 1997; 13: 293–295. [PubMed] [Google Scholar]
2. Weaver IC, Cervoni N, Champagne FA, et al. Эпигенетическое программирование материнским поведением. Nat Neurosci. 2004; 7: 847–854. [PubMed] [Google Scholar]
3. Riggs AD, Xiong Z, Wang L, LeBon JM. Динамика метилирования, эпигенетическая точность и структура Х-хромосомы. Компания Novartis обнаружила симптом. 1998;214:214–25 обсуждение 25–32. [PubMed] [Google Scholar]
4. Ричардс Э.Дж. Унаследованная эпигенетическая изменчивость — пересмотр мягкого наследования. Nat Rev Genet. 2006; 7: 395–401. [PubMed] [Google Scholar]
Nat Rev Genet. 2006; 7: 395–401. [PubMed] [Google Scholar]
5. Kaminsky ZA, Tang T, Wang SC, et al. Профили метилирования ДНК у монозиготных и дизиготных близнецов. Нат Жене. 2009;41:240–245. [PubMed] [Google Scholar]
6. Jaenisch R, Bird A. Эпигенетическая регуляция экспрессии генов, как геном интегрирует внутренние сигналы и сигналы окружающей среды. Нат Жене. 2003; 33 (доп.): 245–254. [PubMed] [Google Scholar]
7. Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Взаимодействия, опосредованные растворителем, в структуре ядра нуклеосомы при разрешении 1,9 а. Дж Мол Биол. 2002; 319:1097–1113. [PubMed] [Google Scholar]
8. Kriaucionis S, Heintz N. Основание ядерной ДНК 5-гидроксиметилцитозин присутствует в нейронах Пуркинье и головном мозге. Наука. 2009; 324:929–930. [Бесплатная статья PMC] [PubMed] [Google Scholar]
9. Маргерон Р., Тройер П., Рейнберг Д. Ключ к развитию, интерпретирующий гистоновый код? Curr Opin Genet Dev. 2005; 15:163–176. [PubMed] [Google Scholar]
2005; 15:163–176. [PubMed] [Google Scholar]
10. Thiagalingam S, Cheng KH, Lee HJ, et al. Гистон деацетилазирует уникальных игроков в формировании эпигенетического гистонового кода. Энн Н.Ю. акад. 2003; 983:84–100. [PubMed] [Google Scholar]
11. Klose RJ, Zhang Y. Регуляция метилирования гистонов путем деметилиминирования и деметилирования. Nat Rev Mol Cell Biol. 2007; 8: 307–318. [PubMed] [Google Scholar]
12. Николсон Т.Б., Чен Т. ЛСД1 деметилирует гистоновые и негистоновые белки. Эпигенетика. 2009; 4:129–132. [PubMed] [Google Scholar]
13. Рейсс Д., Пломин Р., Хетерингтон Э.М. Генетика и психиатрия: неожиданное окно в окружающую среду. Am J Психиатрия. 1991; 148: 283–291. [PubMed] [Google Scholar]
14. Petronis A, Gottesman II, Kan P, et al. Монозиготные близнецы демонстрируют многочисленные эпигенетические различия: ключи к дискордантности близнецов? Шизофр Булл. 2003; 29: 169–178. [PubMed] [Google Scholar]
15. Fraga MF, Ballestar E, Paz MF, et al. Эпигенетические различия возникают в течение жизни монозиготных близнецов. Proc Natl Acad Sci U S A. 2005;102:10604–10609. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Fraga MF, Ballestar E, Paz MF, et al. Эпигенетические различия возникают в течение жизни монозиготных близнецов. Proc Natl Acad Sci U S A. 2005;102:10604–10609. [Бесплатная статья PMC] [PubMed] [Google Scholar]
16. Seeman MV. Психопатология у женщин и мужчин: внимание к женским гормонам. Am J Психиатрия. 1997; 154:1641–1647. [PubMed] [Google Scholar]
17. Auger AP, Jessen HM. Корепрессоры, ядерные рецепторы и эпигенетические факторы на хвосте репрессии ДНК. Психонейроэндокринология. 2009;34 (прил. 1):S39–S47. [Бесплатная статья PMC] [PubMed] [Google Scholar]
18. Каминский З., Ван С.С., Петронис А. Комплексное заболевание, пол и эпигенетика. Энн Мед. 2006; 38: 530–544. [PubMed] [Google Scholar]
19. Ohara K, Xu HD, Mori N, et al. Антиципация и импринтинг при шизофрении. Биол Психиатрия. 1997; 42:760–766. [PubMed] [Google Scholar]
20. Бассет С.С., Аврамопулос Д., Фаллин Д. Доказательства влияния родителя при болезни Альцгеймера с поздним началом. Am J Med Genet. 2002; 114:679–686. [PubMed] [Google Scholar]
Am J Med Genet. 2002; 114:679–686. [PubMed] [Google Scholar]
21. Lamb JA, Barnby G, Bonora E, et al. Анализ локусов предрасположенности к аутизму IMGSAC: доказательства ограниченности пола и специфических эффектов от родителей. J Med Genet. 2005; 42:132–137. [Бесплатная статья PMC] [PubMed] [Google Scholar]
22. Schulze TG, Chen YS, Badner JA, et al. Дополнительные, физически упорядоченные маркеры усиливают сигнал сцепления для биполярного расстройства на хромосоме 18q22. Биол Психиатрия. 2003; 53: 239–243. [PubMed] [Google Scholar]
23. Барлоу Д.П. Гаметический импринтинг у млекопитающих. Наука. 1995; 270:1610–1613. [PubMed] [Google Scholar]
24. Делаваль К., Вагшал А., Фейл Р. Эпигенетическая дерегуляция импринтинга при врожденных заболеваниях аберрантного роста. Биоэссе. 2006; 28: 453–459. [PubMed] [Google Scholar]
25. Петронис А. Происхождение шизофрении: генетический тезис, эпигенетический антитезис и разрешающий синтез. Биол Психиатрия. 2004; 55:965–970. [PubMed] [Google Scholar]
Биол Психиатрия. 2004; 55:965–970. [PubMed] [Google Scholar]
26. Сазерленд Дж. Э., Коста М. Эпигенетика и окружающая среда. Ann NY Acad Sci. 2003; 983:151–160. [PubMed] [Google Scholar]
27. Левенсон Дж.М., Суитт Дж.Д. Эпигенетические механизмы: общая тема формирования памяти у позвоночных и беспозвоночных. Cell Mol Life Sci. 2006; 63:1009–1016. [PubMed] [Google Scholar]
28. Любин Ф.Д., Рот Т.Л., Суитт Д.Д. Эпигенетическая регуляция транскрипции гена BDNF при консолидации памяти о страхе. Дж. Неврологи. 2008; 28:10576–10586. [Бесплатная статья PMC] [PubMed] [Google Scholar]
29. Liu L, van Groen T, Kadish I, Tollefsbol TO. Метилирование ДНК влияет на обучение и память при старении. Нейробиол Старение. 2009;30:549–560. [Бесплатная статья PMC] [PubMed] [Google Scholar]
30. Орландо В. Поликомб, эпигеномы и контроль клеточной идентичности. Сотовый. 2003; 112:599–606. [PubMed] [Google Scholar]
31.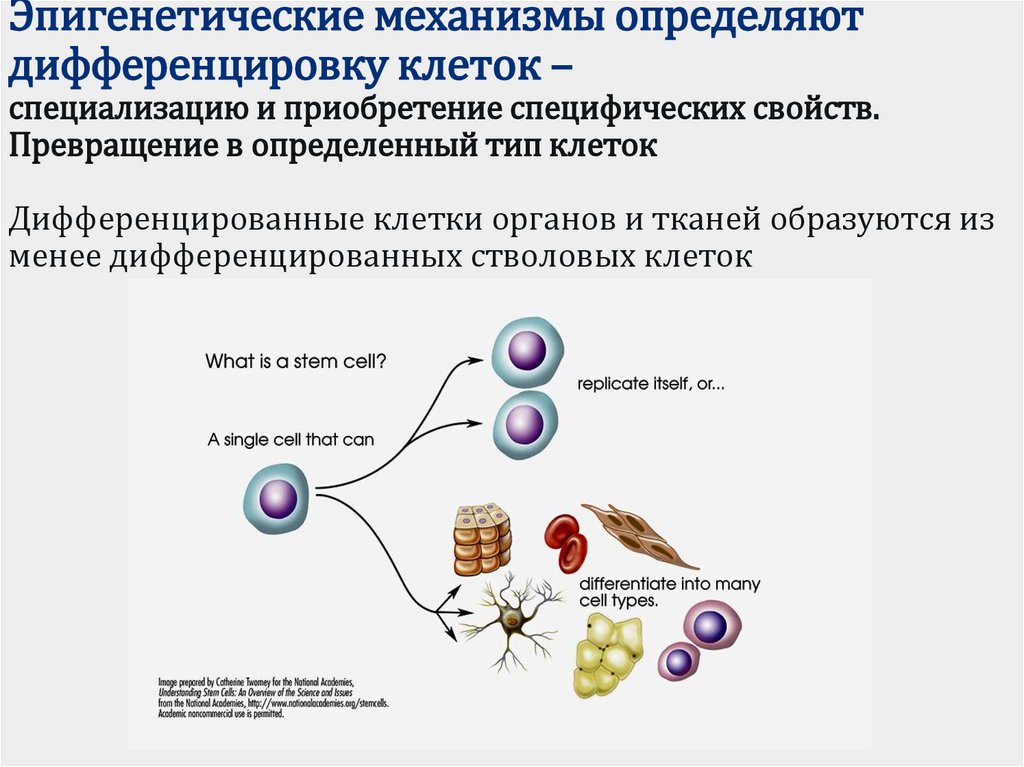 Kim SY, Levenson JM, Korsmeyer S, Sweatt JD, Schumacher A. Онтогенетическая регуляция состава комплекса Eed управляет переключением глобальной модификации гистонов в головном мозге. J Биол. Хим. 2007; 282:9962–9972. [PubMed] [Google Scholar]
Kim SY, Levenson JM, Korsmeyer S, Sweatt JD, Schumacher A. Онтогенетическая регуляция состава комплекса Eed управляет переключением глобальной модификации гистонов в головном мозге. J Биол. Хим. 2007; 282:9962–9972. [PubMed] [Google Scholar]
32. Левенсон Дж. М., О’Риордан К. Дж., Браун К. Д. и соавт. Регуляция ацетилирования гистонов при формировании памяти в гиппокампе. J Biol Chem. 2004; 279:40545–40559. [PubMed] [Google Scholar]
33. Levenson JM, Roth TL, Lubin FD, et al. Доказательства того, что ДНК (цитозин5) метилтрансфераза регулирует синаптическую пластичность в гиппокампе. J Biol Chem. 2006; 281:15763–15773. [PubMed] [Академия Google]
34. Yeh SH, Lin CH, Gean PW. Ацетилирование ядерного фактора-каппаВ в миндалевидном теле крыс улучшает долговременное, но не кратковременное сохранение памяти о страхе. Мол Фармакол. 2004; 65:1286–1292. [PubMed] [Google Scholar]
35. Ивлева Э., Такер Г., Тамминга К.А. Сравнение генов и феноменологии основных психозов шизофрении и биполярного расстройства 1 типа. Шизофр Булл. 2008; 34:734–742. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Шизофр Булл. 2008; 34:734–742. [Бесплатная статья PMC] [PubMed] [Google Scholar]
36. Andreasen NC. Симптомы, признаки и диагностика шизофрении. Ланцет. 1995; 346:477–481. [PubMed] [Google Scholar]
37. Craddock N, O’Donovan MC, Owen MJ. Генетика шизофрении и биполярного расстройства: расслаивающий психоз. J Med Genet. 2005;42:193–204. [Бесплатная статья PMC] [PubMed] [Google Scholar]
38. Крэддок Н., Джонс И. Генетика биполярного расстройства. J Med Genet. 1999; 36: 585–594. [Бесплатная статья PMC] [PubMed] [Google Scholar]
39. Bertelsen A, Gottesman II. Шизоаффективные психозы: генетические ключи к классификации. Am J Med Genet. 1995; 60:7–11. [PubMed] [Google Scholar]
40. Cardno AG, Gottesman II. Близнецовые исследования шизофрении: от соответствия лука и стрелы до звездных войн Mx и функциональной геномики. Am J Med Genet. 2000;97:12–17. [PubMed] [Google Scholar]
41. O’Donovan MC, Craddock NJ, Owen MJ. Генетика психозов; выводы из взглядов на геном. Гум Жене. 2009; 126:3–12. [PubMed] [Google Scholar]
O’Donovan MC, Craddock NJ, Owen MJ. Генетика психозов; выводы из взглядов на геном. Гум Жене. 2009; 126:3–12. [PubMed] [Google Scholar]
42. Mill J, Tang T, Kaminsky Z, et al. Эпигеномное профилирование выявляет изменения метилирования ДНК, связанные с тяжелым психозом. 901:51 Am J Hum Genet. 2008; 82: 696–711. [Бесплатная статья PMC] [PubMed] [Google Scholar]
43. Gupta DS, McCullumsmith RE, Beneyto M, et al. Экспрессия белка метаботропного рецептора глутамата в префронтальной коре и стриатуме при шизофрении. Синапс. 2005; 57: 123–131. [PubMed] [Google Scholar]
44. Иствуд С.Л., Харрисон П.Дж. Снижение экспрессии мРНК везикулярного переносчика глутамата 1 и комплексина II при шизофрении: еще одно свидетельство синаптической патологии, поражающей глутаматные нейроны. Шизофр Рез. 2005; 73: 159–172. [PubMed] [Google Scholar]
45. Брюно Э.Г., Маккалламсмит Р.Э., Харутюнян В., Дэвис К.Л., Меадор Вудрафф Дж.Х. Повышенная экспрессия мРНК глутаминазы и глутаминсинтетазы в таламусе при шизофрении. Шизофр Рез. 2005;75:27–34. [PubMed] [Google Scholar]
Шизофр Рез. 2005;75:27–34. [PubMed] [Google Scholar]
46. Мияока Т., Сено Х., Ишино Х. Повышенная экспрессия Wnt-1 в мозге шизофреников. Шизофр Рез. 1999; 38:1–6. [PubMed] [Академия Google]
47. Grayson DR, Jia X, Chen Y, et al. Гиперметилирование промотора рилина при шизофрении. Proc Natl Acad Sci USA. 2005; 102:9341–9346. [Бесплатная статья PMC] [PubMed] [Google Scholar]
48. Tochigi M, Iwamoto K, Bundo M, et al. Статус метилирования промоутерной области рилина в головном мозге больных шизофренией. Биол Психиатрия. 2008; 63: 530–533. [PubMed] [Google Scholar]
49. Буллок В.М., Кардон К., Бустильо Дж., Робертс Р.С., Перроне-Биззозеро Н.И. Измененная экспрессия генов, участвующих в ГАМКергической передаче и нейромодуляции активности гранулярных клеток в мозжечке больных шизофренией. Am J Психиатрия. 2008; 165:1594–1603. [PubMed] [Google Scholar]
50. Volk DW, Lewis DA. Нарушение префронтального торможения при шизофрении: значение для когнитивной дисфункции. Физиол Поведение. 2002; 77: 501–505. [PubMed] [Google Scholar]
Физиол Поведение. 2002; 77: 501–505. [PubMed] [Google Scholar]
51. Hashimoto T, Volk DW, Eggan SM, et al. Дефицит экспрессии генов в подклассе нейронов ГАМК в префронтальной коре у больных шизофренией. Дж. Неврологи. 2003; 23:6315–63126. [Бесплатная статья PMC] [PubMed] [Google Scholar]
52. Huang HS, Matevossian A, Whittle C, et al. Префронтальная дисфункция при шизофрении включает регулируемое лейкемией 1 смешанного происхождения метилирование гистонов на промоторах ГАМКергических генов. Дж. Неврологи. 2007; 27:11254–11262. [Бесплатная статья PMC] [PubMed] [Google Scholar]
53. Шарма Р.П., Грейсон Д.Р., Гэвин Д.П. Экспрессия гистондеактилазы 1 повышена в префронтальной коре у больных шизофренией: анализ коллекции микрочипов Национального банка данных о мозге. Шизофр Рез. 2008; 98:111–117. [Бесплатная статья PMC] [PubMed] [Google Scholar]
54. Huang HS, Akbarian S. Экспрессия мРНК GAD1 и метилирование ДНК в префронтальной коре у пациентов с шизофренией. PLoS Один. 2007;2:e809. [Бесплатная статья PMC] [PubMed] [Google Scholar]
PLoS Один. 2007;2:e809. [Бесплатная статья PMC] [PubMed] [Google Scholar]
55. Akbarian S, Ruehl MG, Bliven E, et al. Изменения хроматина, связанные со сниженной экспрессией метаболических генов в префронтальной коре у больных шизофренией. Arch Gen Психиатрия. 2005; 62:829–840. [PubMed] [Google Scholar]
56. Veldic M, Caruncho HJ, Liu WS, et al. мРНК ДНК-метилтрансферазы 1 избирательно сверхэкспрессируется в телэнцефалических ГАМКергических интернейронах головного мозга больных шизофренией. Proc Natl Acad Sci U S A. 2004;101:348–353. [Бесплатная статья PMC] [PubMed] [Google Scholar]
57. Veldic M, Kadriu B, Maloku E, et al. Эпигенетические механизмы, выраженные в ГАМКергических нейронах базальных ганглиев, позволяют дифференцировать шизофрению от биполярного расстройства. Шизофр Рез. 2007; 91:51–61. [Бесплатная статья PMC] [PubMed] [Google Scholar]
58. Zhubi A, Veldic M, Puri NV, et al. Активация ДНК-метилтрансферазы 1 и 3а, экспрессируемая в теленцефалических ГАМКергических нейронах больных шизофренией, также обнаруживается в лимфоцитах периферической крови. Шизофр Рез. 2009; 111:115–122. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Шизофр Рез. 2009; 111:115–122. [Бесплатная статья PMC] [PubMed] [Google Scholar]
59. Satta R, Maloku E, Zhubi A, et al. Никотин снижает экспрессию ДНК-метилтрансферазы 1 и метилирование промотора декарбоксилазы глутаминовой кислоты 67 в ГАМКергических интернейронах. Proc Natl Acad Sci U S A. 2008;105:16356–16361. [Бесплатная статья PMC] [PubMed] [Google Scholar]
60. Lising-Enriquez K, George TP. Лечение сопутствующего употребления табака у людей с серьезными психическими заболеваниями. J Неврологическая психиатрия. 2009;34:E1–E2. [Бесплатная статья PMC] [PubMed] [Google Scholar]
61. McGowan PO, Kato T. Эпигенетика при расстройствах настроения. Environment Health Prev Med. 2008; 13:16–24. [Бесплатная статья PMC] [PubMed] [Google Scholar]
62. Kuratomi G, Iwamoto K, Bundo M, et al. Аберрантное метилирование ДНК, связанное с биполярным расстройством, выявлено у дискордантных монозиготных близнецов. Мол Психиатрия. 2008; 13:429–441. [PubMed] [Google Scholar]
2008; 13:429–441. [PubMed] [Google Scholar]
63. Meyer J, Johannssen K, Freitag CM, et al. Редкие варианты гена, кодирующего котранспортер хлорида калия 3, связаны с биполярным расстройством. Int J Neuropsychopharmacol. 2005; 8: 495–504. [PubMed] [Google Scholar]
64. Moser D, Ekawardhani S, Kumsta R, et al. Функциональный анализ полиморфизма промотора котранспортера 3 хлорида калия (SLC12A6), приводящего к дополнительному сайту метилирования ДНК. Нейропсихофармакология. 2009; 34:458–467. [PubMed] [Google Scholar]
65. Uyanik G, Elcioglu N, Penzien J, et al. Новые укороченные и миссенс-мутации гена KCC3, связанные с синдромом Андермана. Неврология. 2006;66:1044–1048. [PubMed] [Google Scholar]
66. Sennvik K, Fastbom J, Blomberg M, et al. Уровни белка-предшественника амилоида, расщепленного альфа- и бета-секретазой, в спинномозговой жидкости пациентов с болезнью Альцгеймера. Neurosci Lett. 2000; 278:169–172. [PubMed] [Google Scholar]
[PubMed] [Google Scholar]
67. Kim J, Basak JM, Holtzman DM. Роль аполипопротеина Е в развитии болезни Альцгеймера. Нейрон. 2009;63:287–303. [Бесплатная статья PMC] [PubMed] [Google Scholar]
68. Liddell MB, Lovestone S, Owen MJ. Генетический риск болезни Альцгеймера, консультирование родственников. Бр Ж Психиатрия. 2001; 178:7–11. [PubMed] [Google Scholar]
69. Manzano S, Gonzalez J, Marcos A, Matias-Guiu J. [Генетика и болезнь Альцгеймера]. Неврология. 2009; 24:83–89. [PubMed] [Google Scholar]
70. Грюнблатт Э. Общие черты генетики болезни Альцгеймера и болезни Паркинсона. Эксперт преподобный Нейротер. 2008; 8: 1865–1877. [PubMed] [Google Scholar]
71. Gatz M, Reynolds CA, Fratiglioni L, et al. Роль генов и окружающей среды в объяснении болезни Альцгеймера. Arch Gen Психиатрия. 2006; 63: 168–174. [PubMed] [Google Scholar]
72. Bassett SS, Avramopoulos D, Perry RT, et al. Еще одно свидетельство материнского родительского влияния на хромосому 10 при позднем начале болезни Альцгеймера. Am J Med Genet B Neuropsychiatr Genet. 2006; 141B: 537–540. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Am J Med Genet B Neuropsychiatr Genet. 2006; 141B: 537–540. [Бесплатная статья PMC] [PubMed] [Google Scholar]
73. Dolinoy DC, Das R, Weidman JR, Jirtle RL. Метастабильные эпиаллели, импринтинг и фетальное происхождение болезней взрослых. Педиатр Рез. 2007; 61:30R–37R. [PubMed] [Google Scholar]
74. Siegmund KD, Connor CM, Campan M, et al. Метилирование ДНК в коре головного мозга человека динамически регулируется на протяжении всей жизни и затрагивает дифференцированные нейроны. PLoS Один. 2007;2:e895. [Бесплатная статья PMC] [PubMed] [Google Scholar]
75. Hoyaux D, Decaestecker C, Heizmann CW, et al. Белки S100 в Corpora amylacea из нормального человеческого мозга. Мозг Res. 2000;867:280–288. [PubMed] [Google Scholar]
76. Wang SC, Oelze B, Schumacher A. Возрастной эпигенетический дрейф при болезни Альцгеймера с поздним началом. PLoS Один. 2008;3:e2698. [Бесплатная статья PMC] [PubMed] [Google Scholar]
77. Bjornsson HT, Sigurdsson MI, Fallin MD, et al. Внутрииндивидуальное изменение метилирования ДНК с течением времени с семейной кластеризацией. ЯМА. 2008; 299:2877–83. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Bjornsson HT, Sigurdsson MI, Fallin MD, et al. Внутрииндивидуальное изменение метилирования ДНК с течением времени с семейной кластеризацией. ЯМА. 2008; 299:2877–83. [Бесплатная статья PMC] [PubMed] [Google Scholar]
78. Bollati V, Schwartz J, Wright R, et al. Снижение метилирования геномной ДНК в результате старения в когорте пожилых людей. Старение меха Dev. 2009; 130:234–239. [Бесплатная статья PMC] [PubMed] [Google Scholar]
79. Kumbier E, Domes G, Herpertz-Dahlmann B, Herpertz SC. [Аутизм и аутистические расстройства: исторические и современные аспекты]. Нервенарцт. 2010;81:55–65. [PubMed] [Академия Google]
80. Hallmayer J, Glasson EJ, Bower C, et al. О двойном риске при аутизме. Am J Hum Genet. 2002; 71:941–946. [Бесплатная статья PMC] [PubMed] [Google Scholar]
81. Пиггот Дж., Ширинян Д., Шеммассян С., Вазириан С., Аларкон М. Подходы нейронных систем к нейрогенетике расстройств аутистического спектра. Неврология. 2009; 164: 247–256. [PubMed] [Google Scholar]
2009; 164: 247–256. [PubMed] [Google Scholar]
82. Crespi B. Геномный импринтинг в развитии и эволюции состояний психотического спектра. Biol Rev Camb Philos Soc. 2008; 83: 441–493. [PubMed] [Google Scholar]
83. Yusufzai TM, Wolffe AP. Функциональные последствия мутаций синдрома Ретта на MeCP2 человека. Рез. нуклеиновых кислот. 2000;28:4172–4179. [Бесплатная статья PMC] [PubMed] [Google Scholar]
84. Young JI, Hong EP, Castle JC, et al. Регуляция сплайсинга РНК метил-зависимым репрессором транскрипции метил-CpG-связывающим белком 2. Proc Natl Acad Sci U S A. 2005;102:17551–17558. [Бесплатная статья PMC] [PubMed] [Google Scholar]
85. Kriaucionis S, Bird A. Метилирование ДНК и синдром Ретта. Хум Мол Жене. 2003;12 Спец. № 2:R221–R227. [PubMed] [Google Scholar]
86. Guy J, Hendrich B, Holmes M, Martin JE, Bird A. Нулевая мутация Mecp2 мыши вызывает неврологические симптомы, имитирующие синдром Ретта. Нат Жене. 2001; 27:322–326. [PubMed] [Google Scholar]
Нат Жене. 2001; 27:322–326. [PubMed] [Google Scholar]
87. Collins AL, Levenson JM, Vilaythong AP, et al. Умеренная сверхэкспрессия MeCP2 вызывает прогрессирующее неврологическое расстройство у мышей. Гум Мол Генет. 2004; 13:2679–2689. [PubMed] [Google Scholar]
88. Shahbazian M, Young J, Yuva-Paylor L, et al. Мыши с укороченным MeCP2 повторяют многие признаки синдрома Ретта и обнаруживают гиперацетилирование гистона h4. Нейрон. 2002; 35: 243–254. [PubMed] [Google Scholar]
89. Маэдзава И., Сванберг С., Харви Д., ЛаСалле Дж. М., Джин Л. В. Астроциты синдрома Ретта являются аномальными и распространяют дефицит MeCP2 через щелевые контакты. Дж. Неврологи. 2009;29: 5051–5061. [Бесплатная статья PMC] [PubMed] [Google Scholar]
90. Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Снижение экспрессии MeCP2 часто встречается в лобной коре аутизма и коррелирует с аберрантным метилированием промотора MECP2. Эпигенетика. 2006; 1:e1–e11. [Бесплатная статья PMC] [PubMed] [Google Scholar]
2006; 1:e1–e11. [Бесплатная статья PMC] [PubMed] [Google Scholar]
91. Nagarajan RP, Patzel KA, Martin M, et al. Метилирование промотора MECP2 и инактивация Х-хромосомы при аутизме. Аутизм Res. 2008; 1:169–178. [Бесплатная статья PMC] [PubMed] [Google Scholar]
92. Allan AM, Liang X, Luo Y, et al. Потеря белка 1, связывающего метил-CpG, приводит к поведенческим нарушениям, подобным аутизму. Хум Мол Жене. 2008;17:2047–2057. [Бесплатная статья PMC] [PubMed] [Google Scholar]
93. Адегбола А., Гао Х., Соммер С., Браунинг М. Новая мутация в JARID1C/SMCX у пациента с расстройством аутистического спектра (РАС). Am J Med Genet A . 2008; 146А: 505–511. [PubMed] [Google Scholar]
94. Curran LK, Newschaffer CJ, Lee LC, et al. Поведение, связанное с лихорадкой у детей с расстройствами аутистического спектра. Педиатрия. 2007; 120:e1386–e1392. [PubMed] [Google Scholar]
95. Plagge A, Isles AR, Gordon E, et al. Импринтированный Nesp55 влияет на поведенческую реакцию на новую среду. Мол клеточный биол. 2005; 25:3019–3026. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Мол клеточный биол. 2005; 25:3019–3026. [Бесплатная статья PMC] [PubMed] [Google Scholar]
96. Mehler MF, Purpura DP. Аутизм, лихорадка, эпигенетика и голубое пятно. Brain Res Rev. 2009; 59: 388–392. [Бесплатная статья PMC] [PubMed] [Google Scholar]
97. Kundakovic M, Chen Y, Costa E, Grayson DR. Ингибиторы ДНК-метилтрансферазы координированно индуцируют экспрессию генов человеческого рилина и декарбоксилазы глутаминовой кислоты 67. Мол Фармакол. 2007; 71: 644–653. [PubMed] [Google Scholar]
98. Dash PK, Orsi SA, Moore AN. Ингибирование гистондеактилазы в сочетании с поведенческой терапией улучшает обучение и память после черепно-мозговой травмы. Неврология. 2009; 213:47–59. [Бесплатная статья PMC] [PubMed] [Google Scholar]
99. Hockly E, Richon VM, Woodman B, et al. Субероланилид гидроксамовой кислоты, ингибитор гистоновой деацетилазы, улучшает двигательные расстройства у мышиной модели болезни Гентингтона. Proc Natl Acad Sci U S A. 2003;100:2041–2046. [Бесплатная статья PMC] [PubMed] [Google Scholar]
2003;100:2041–2046. [Бесплатная статья PMC] [PubMed] [Google Scholar]
100. Camelo S, Iglesias AH, Hwang D, et al. Транскрипционная терапия ингибитором гистоновой деацетилазы трихостатином А облегчает течение экспериментального аутоиммунного энцефаломиелита. J Нейроиммунол. 2005; 164:10–21. [PubMed] [Google Scholar]
101. Chen PS, Peng GS, Li G, et al. Вальпроат защищает дофаминергические нейроны в культурах нейронов/глии среднего мозга, стимулируя высвобождение нейротрофических факторов из астроцитов. Мол Психиатрия. 2006; 11:1116–1125. [PubMed] [Google Scholar]
102. Дагтас А.С., Эденс Э.Р., Гилберт К.М. Ингибитор гистондеацетилазы использует p21(Cip1) для поддержания анергии в CD4(+) Т-клетках. Int Immunopharmacol. 2009; 9:1289–1297. [PubMed] [Google Scholar]
103. Langley B, D’Annibale MA, Suh K, et al. Импульсное ингибирование гистоновых деацетилаз индуцирует полную устойчивость к окислительной гибели нейронов коры без токсичности и выявляет роль цитоплазматического p21 (waf1/cip1) в независимой от клеточного цикла нейропротекции. Дж. Неврологи. 2008; 28:163–176. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Дж. Неврологи. 2008; 28:163–176. [Бесплатная статья PMC] [PubMed] [Google Scholar]
104. Gilad R, Izkovitz N, Dabby R, et al. Лечение эпилептического статуса и острых повторяющихся припадков в/в. вальпроевая кислота против фенитоина. Акта Нейрол Сканд. 2008; 118:296–300. [PubMed] [Google Scholar]
105. Bowden CL. Спектр эффективности вальпроата в нейропсихиатрии. Опыт Преподобный Нейротер. 2007;7:9–16. [PubMed] [Google Scholar]
106. Sajatovic M, Coconcea N, Ignacio RV, et al. Дополнительный пролонгированный вальпроат полунатрий при шизофрении в позднем возрасте. Int J Гериатрическая психиатрия. 2008; 23:142–147. [PubMed] [Google Scholar]
107. Хайдук П. Дж., Грир Дж. Десятилетие разработки лекарств на основе фрагментов: стратегические достижения и извлеченные уроки. Nat Rev Drug Discov. 2007: 6211–219. [PubMed] [Google Scholar]
108. Штраусберг Р.Л., Шрайбер С.Л. От знания к контролю: путь от геномики к лекарствам с использованием низкомолекулярных зондов. Наука. 2003; 300:294–295. [PubMed] [Google Scholar]
Наука. 2003; 300:294–295. [PubMed] [Google Scholar]
109. Kubicek S, O’Sullivan RJ, August EM, et al. Разворот h4K9me2 низкомолекулярным ингибитором гистонметилтрансферазы G9a. молярная ячейка. 2007; 25: 473–481. [PubMed] [Google Scholar]
110. Вагонер Д. Механизмы болезни: эпигенез. Семин Педиатр Нейрол. 2007; 14:7–14. [PubMed] [Google Scholar]
111. Пушпарадж П.Н., Мелендес А.Дж. Короткие интерферирующие РНК (киРНК) как новый терапевтический препарат. Clin Exp Pharmacol Physiol. 2006; 33: 504–510. [PubMed] [Google Scholar]
112. Bernstein E, Allis CD. РНК встречается с хроматином. Гены Dev. 2005;19:1635–1655. [PubMed] [Google Scholar]
113. Джеймисон А.С., Миллер Дж.К., Пабо К.О. Открытие лекарств с помощью инженерных белков цинковых пальцев. Nat Rev Drug Discov. 2003; 2: 361–368. [PubMed] [Google Scholar]
114. Xu GL, Bestor TH. Метилирование цитозина направлено на заранее определенные последовательности. Нат Жене. 1997; 17:376–378. [PubMed] [Google Scholar]
Нат Жене. 1997; 17:376–378. [PubMed] [Google Scholar]
115. Roelfsema JH, White SJ, Ariyurek Y, et al. Генетическая гетерогенность при синдроме Рубинштейна-Тайби: мутации как в генах CBP, так и в генах EP300 вызывают заболевание. 901:51 Am J Hum Genet. 2005; 76: 572–580. [PMC free article] [PubMed] [Google Scholar]
116. Philibert RA, Gunter TD, Beach SR, Brody GH, Madan A. Метилирование MAOA связано с никотиновой и алкогольной зависимостью у женщин. Am J Med Genet B Neuropsychiatr Genet. 2008; 147B: 565–570. [Бесплатная статья PMC] [PubMed] [Google Scholar]
117. Bonsch D, Lenz B, Fiszer R, et al. Пониженная экспрессия мРНК ДНК-метилтрансферазы (DNMT3b) связана с гиперметилированием геномной ДНК у пациентов с хроническим алкоголизмом. J Нейронная передача. 2006; 113:1 299–1304. [PubMed] [Google Scholar]
118. Stack EC, Del Signore SJ, Luthi-Carter R, et al. Модуляция динамики нуклеосом при болезни Гентингтона. Хум Мол Жене.